




-
竹林金针虫是取食竹笋的叩甲幼虫的总称,虫害发生时竹笋产量显著减少,严重时竹种无法成竹,制约了竹林的自然更新[1-2]。筛胸梳爪叩甲Melanotus cribricollis隶属于鞘翅目Coleoptera叩甲科 Elateridae梳爪叩甲属Melanotus,其幼虫为中国南方早园竹Phyllostachys propinqua竹林的优势种。化学药剂防治竹林金针虫易造成环境污染、抗药性和食品安全等问题,阻碍以“生态、健康、有机”为标签的林产品产业发展,因此近年来逐渐采用生物技术防治该虫害。绿僵菌Metarhizium sp.是常见的昆虫致病真菌[3-4],对鞘翅目和鳞翅目Lepidoptera等害虫均有一定防效[5-6]。研究竹林金针虫抗绿僵菌相关基因的表达有利于深入了解害虫免疫抗性机制和虫菌互作机制,也有利于生防菌菌株改造等研究,提高其对农林害虫防治的效果。实时荧光定量聚合酶链式反应(qRT-PCR)是研究基因表达分析的关键技术,省时、高效、应用广泛;为保证目的基因定量表达结果的准确性,需引入表达稳定的内参基因作为参照[7]。内参基因又称看家基因,理想的内参基因其表达水平应在各条件下保持稳定,而实际上不同条件下许多内参基因的表达并不稳定[8-9]。因此,即使是同一物种,不同试验条件下的内参基因也不能一概而用,qRT-PCR试验之前应对所选内参基因的表达稳定性进行评估。常用的qRT-PCR内参基因包括ACT (肌动蛋白)、TUB (微管蛋白)、GAPDH (甘油醛-3-磷酸脱氢酶)、UBC (泛素结合酶)、18S rRNA (核糖体蛋白S18)、28S rRNA (核糖体蛋白S28)、EF1 (延伸因子1)、SYN1 (突触融合蛋白1)、SYN6 (突触融合蛋白6)、RPS3 (核糖体蛋白S3)、PRS20 (泛素-核糖体蛋白S20)、PRS27a (泛素-核糖体蛋白S27a)、RPL10 (核糖体蛋白L10)、RPL13α (核糖体蛋白S13α)、AK (精氨酸激酶)和V-ATPase (液泡型ATP合酶)等。符伟等[10]分析了小菜蛾Plutella xylostella候选内参基因在Bt毒素诱导时的表达稳定性,最终筛选出由PRS13、RPL32和 EF1α组成的最佳内参基因组合。冯波等[11] 筛选出最适合校正松墨天牛Monochamus alternatus化学感受组织基因表达的GAPDH和TUB内参基因组合。刘金泊[12]评价了赤拟谷盗Tribolium castaneum的候选内参基因在磷化氢诱导条件下的表达稳定性,筛选出RPS18和 RPL13α最佳内参基因组合。杨苓等[13]筛选出了适合桃蛀螟Conogethes punctiferalis不同发育时期和不同组织的基因表达研究的2组最佳内参组合(RP49和GAPDH,RPL13和RP49)。陶蓉等[14]则分别筛选出了美国白蛾Hyphantria cunea不同发育阶段、不同温度和不同组织的3组最佳内参基因(RPL12和EF1β,EF1α和GAPDH,ACT和RPS16)。目前,关于筛胸梳爪叩甲幼虫内参基因的筛选评估尚未见报道。本研究对平沙绿僵菌Metarhizium pingshaense侵染不同时期的筛胸梳爪叩甲幼虫qRT-PCR内参基因进行了筛选和评估,以期选出最佳的内参基因,为今后开展竹林金针虫等昆虫的基因表达研究提供参考。
-
供试筛胸梳爪叩甲幼虫于2020年3月下旬至4月中旬采自浙江省湖州市德清县的早园竹林地,带回室内置于塑料养虫箱中,箱内装满约3/4的土壤(湿度为10% ± 1%),定期喂以鲜笋。供试平沙绿僵菌WP08菌株由中国林业科学研究院亚热带林业研究所森林保护团队提供,该菌株最初从僵死的筛胸梳爪叩甲幼虫虫体上分离获得[15]。WP08菌株接种于PDA培养基,产孢3周左右收集分生孢子。供试土壤采自德清早园竹园区,40 目的网筛过筛,高温灭菌(121 ℃、1 h),烘干备用。
-
取一定量的平沙绿僵菌WP08分生孢子与供试土壤,混匀后调节土壤水分为(10 ± 1)%,土壤孢子密度为1.1×107个·g−1。挑选活力强且个体大小一致的幼虫置于处理样品中,培养至7、12和17 d时取样,以未经绿僵菌侵染处理的幼虫作为对照组(0 d),随机取样5头·份−1。试验在人工气候培养箱中进行,温度设定为(25 ± 1) ℃,空气相对湿度为(80 ± 10)%,黑暗条件,设置3个生物学重复。所有样品用无菌水清洗数遍,用体积分数75%的乙醇表面消毒后,再用无菌水清洗1遍;无菌滤纸吸干虫体体表水分,液氮速冻,存放于−80 ℃备用。
-
利用景新微量动物组织总RNA提取试剂盒(SIMGEN,杭州)提取不同样品的RNA,具体操作参照试剂盒说明书。利用质量浓度1.0%的琼脂糖凝胶电泳和紫外分光光度计(美国Thermo Scientific)检测所提取总RNA的质量和质量浓度。利用无菌双蒸水(ddH2O)将所有样品RNA稀释到125 mg·L−1备用。按照PrimeScriptTM Reagent kit with gDNA Eraser(Perfect Real Time)反转录试剂盒(Takara)说明书合成样品cDNA,每个样品取7.0 μL RNA。
-
参考昆虫常用内参基因,根据筛胸梳爪叩甲幼虫的转录组测序注释结果[16],筛选不同内参基因(β-actin、α-tubulin、GAPDH、AK、SYN6、RPS3、RPL13α、RPL10、RPS27a、RPL32、RPS20和EF-1α)的同源序列,同时筛选昆虫免疫应答相关基因的6个同源序列[PGRP、Prx、SDR(1)、SDR(2)、SDR(3)和SDR(4)],并利用在线网站http://bioinfo.ut.ee/primer3-0.4.0/针对基因开放阅读框(ORF)区设计qRT-PCR扩增引物。所有引物均由北京擎科新业生物技术有限公司合成。
以cDNA(0 d)为模板进行普通PCR扩增,并根据电泳结果筛选出扩增条带单一的特异性引物对。PCR扩增反应体系为:2×TSINGKE Master Mix 10.0 μL,上下游引物(10 μmol·L−1)各1.5 μL,模板cDNA 3.0 μL,ddH2O补足至20.0 μL。扩增程序为:94 ℃预变性5 min;94 ℃变性30 s,退火55 ℃,72 ℃延伸2 min,共35个循环;最后于72 ℃补平7 min,终止温度为4 ℃。PCR扩增反应在LifeECO基因扩增仪(杭州博日)上进行。取PCR扩增产物3.0 μL,点样于质量浓度为1.5%的琼脂糖凝胶上,TS-GelRed核酸染料染色,1×TAE缓冲液中电泳30~40 min(120 V),利用Bio-Rad凝胶成像分析仪(美国伯乐)摄取扩增图谱。
-
标准曲线构建。将反转录获得的cDNA(0 d)稀释2倍后,再依次稀释5个梯度,各梯度依次稀释5倍。以梯度稀释的cDNA为模板进行qRT-PCR,反应在Line Gene 9600 Plus实时荧光定量PCR仪(杭州博日)上进行,使用Gene-9660 software采集数据。PCR扩增反应体系为:2×SYBR Green Mix 5.0 μL,上下游引物(5 μmol·L−1)各0.2 μL,模板cDNA 1.0 μL,ddH2O补足至10.0 μL。qRT-PCR扩增程序:95 ℃预变性2 min;95 ℃变性15 s,60 ℃退火和延伸1 min,共40个循环,60 ℃采集荧光信号。扩增结束后做熔解曲线分析以检测扩增产物的特异性。熔解曲线程序:95 ℃,15 s;65 ℃,1 min;95 ℃,20 s,隔20 s步进0.2 ℃;30 ℃,1 min。各样品进行3次技术重复,取循环阈值(cycle threshold,Ct)的平均值。判断每对引物是否具有特异性以及是否有引物二聚体,熔解曲线为单峰时用于后续分析。
扩增效率分析。利用SPSS 19.0软件分析cDNA模板质量浓度(对数值)和Ct值的线性关系,获得相关系数(R2)和斜率(S),利用公式E=(10−1/S−1)×100%计算引物的扩增效率[17]。
-
以0(ck)、7、12和17 d取样的cDNA为模板,利用1.4节筛选出的引物对进行qRT-PCR,具体程序同1.4节所述。利用Excel 2003,GeNorm[18]、Normfinder[19]和Bestkeeper[20]软件根据Ct平均值分析候选内参基因的表达稳定性。GeNorm根据表达稳定度(M值)大小对候选内参基因进行排序,M值越小表示基因表达越稳定;根据标准化因子的配对差异分析(Vn/n+1)判定最佳的内参基因数组,Vn/n+1<0.15代表该条件下内参基因最佳数目为n个。Bestkeeper软件通过标准差(SD)和概率值(P)直接分析Ct值,SD>1或者P>0.05时,判定该基因不适合作为内参基因。具体操作方法参考文献[21]。
-
选择免疫应答相关基因的6个同源序列作为目的基因,分析1.6节选出的内参基因在不同处理间(0、7、12和17 d)的相对表达量变化,以验证其表达稳定性。目的基因相对表达量的计算采用
${2^{{\rm{ - }}\Delta \Delta {C_{\rm{t}}}}} $ 法。其中:ΔΔCt=ΔCt-实验−ΔCt-对照,△Ct=Ct-目的基因−Ct-内参基因。采用统计学单因素方差分析及最小显著性差异法(LSD)多重比较分析差异显著性。 -
由图1可见:所有样品D(260)/D(280)均为1.8~2.1,凝胶电泳结果可见2条清晰条带(28S和18S rRNA),说明总RNA较为完整,未出现明显降解。
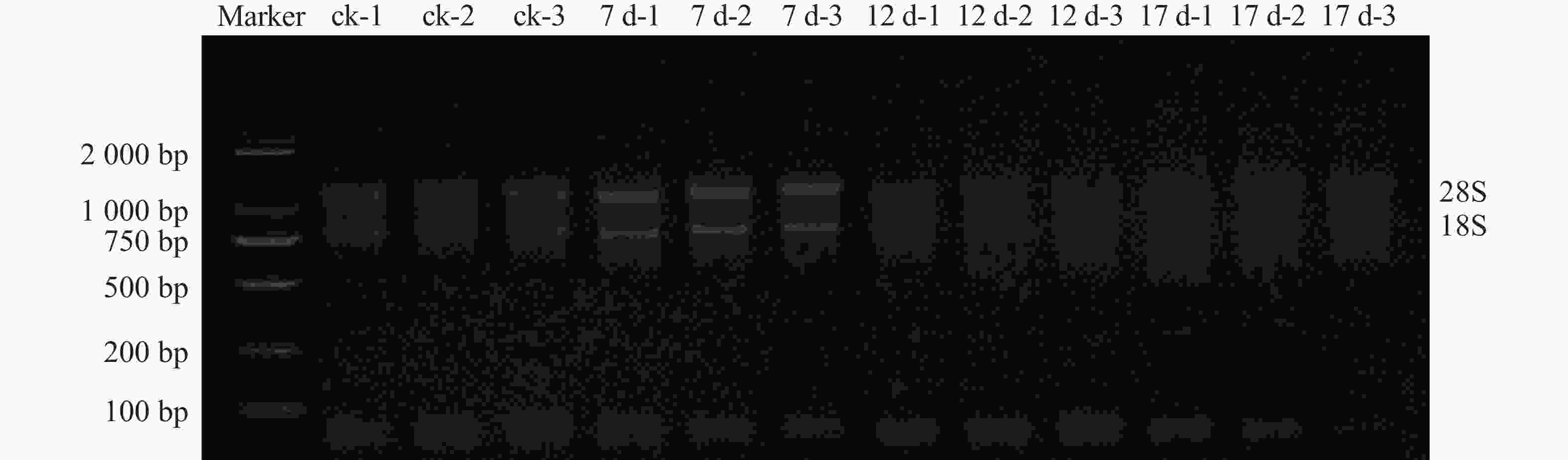
图 1 总RNA电泳图
Figure 1. Electrophoretogram of total RNA
-
根据筛胸梳爪叩甲幼虫的转录组测序数据,筛选出24条候选内参基因和6条目的基因的同源序列,根据普通PCR扩增结果选出条带单一的22对特异性引物,再根据qRT-PCR的引物熔解曲线、R2和扩增效率进一步筛选出6对内参基因(β-actin、GAPDH、α-tubulin、RPL13α、RPS27a和RPS3)和6对目的基因的引物。12对引物的熔解曲线均为单峰,R2为0.989~0.998,扩增效率为88.69%~112.48%,表明引物具有特异性,cDNA的质量浓度和Ct值存在明确的线性关系,引物扩增效率符合荧光定量要求(表1)。
表 1 qRT-PCR引物信息
Table 1. Informations of qRT-PCR primers
基因 引物序列 产物长度/bp 扩增效率/% R2 β-actin F:GGATACCTCTTTTGCTCTGGG,R:ATCAGGGTGTCATGGTTGG 75 112.48 0.993 GAPDH F:CTACTCATGGTCGTTACAAGGG,R:TTCTACAACGTATTCAGCTCCAG 140 101.79 0.993 α-tubulin F:GAAGCTCGTGAAGATTTGGC,R:ACCTTCGCCTTCTCCTTCTC 137 100.63 0.996 RPL13α F:CTGAGGAAGAGCGTAAGGTG,R:TCAGCACGAGCCTTTCTTAAG 145 106.97 0.990 RPS27a F:CTTGTCCTGAATCTTTGCCTTG,R:GTTCTTTTGGTAGCGTGTCATG 146 96.20 0.998 RPS3 F:CAATAGCGCACAAACCACG,R:TGTATTGGGTGAAAAGGGAAGG 128 111.93 0.992 PGRP F:TGTCGTACTTCTGGCTATCATTG,R:TGTGATGGAGGGTTTACTTGC 123 88.69 0.989 Prx F:CTATCCCTTAGACTTCACCTTCG,R:ATTTCTCCCAAACCTCCCTG 171 106.67 0.996 SDR(1) F:CGGCATTGACGGAAACTTTAC,R:GGTTTCCACAGACTTTTGCG 126 109.16 0.995 SDR(2) F:AGGTGCTAGTTCGGGAATTG,R:CGTGTAATTTGCCAGGTTTTCC 140 99.49 0.997 SDR(3) F:GGATTACGAGCATAAGTCCTGG,R:CGGCGATGTCTTCAGATTTTAAC 126 106.58 0.996 SDR(4) F:TTAGGGTTTCAGTCAAGGCAG,R:GAAGCCGTCCAAGATATGAAAG 148 93.34 0.994 -
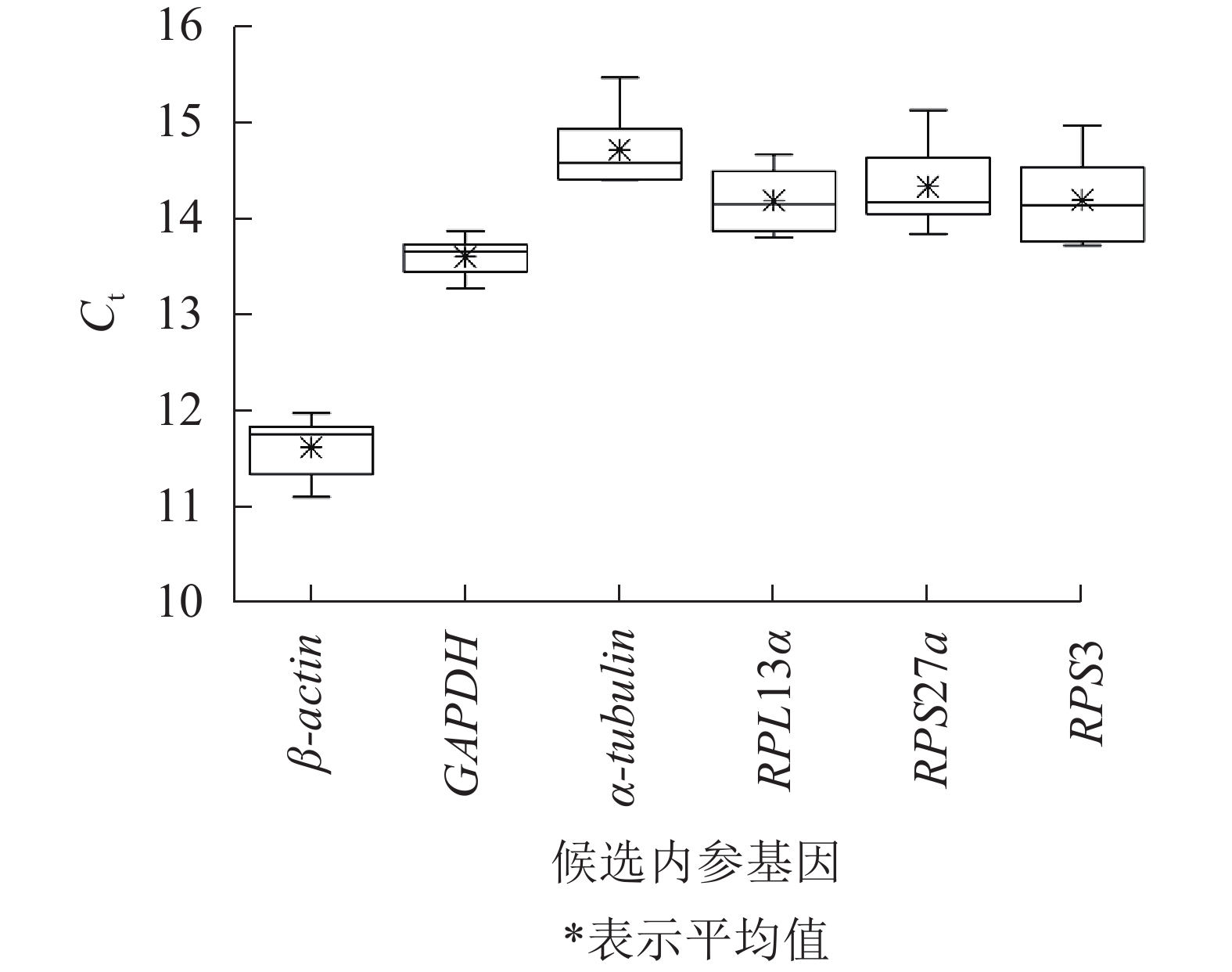
不同平沙绿僵菌侵染处理时长(0、7、12和17 d)下,筛胸梳爪叩甲幼虫6个候选内参基因的Ct值为11.110~15.463(图2),表明本试验条件下所选内参基因表达水平较高。但不同基因的Ct值存在差异,其中β-actin最低,平均Ct值为11.622;α-tubulin最高,平均Ct值为14.712;GAPDH的Ct值跨度最小(13.273~13.873);RPS3跨度最大(13.720~14.970)。
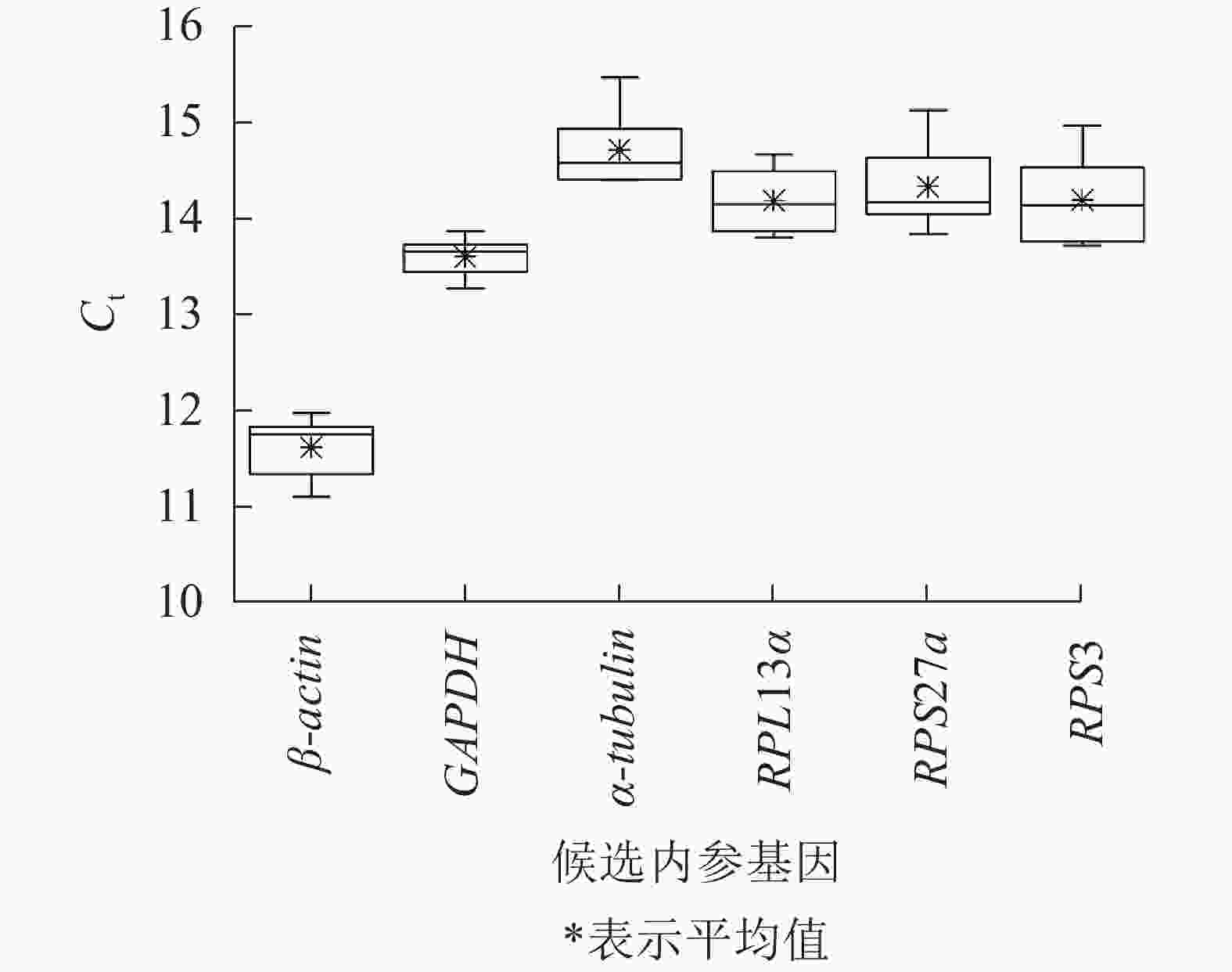
图 2 筛胸梳爪叩甲幼虫候选内参基因的Ct值分析
Figure 2. Ct value analysis of candidate reference genes of Melanotus cribricollis larvae
-
GeNorm软件计算基因的表达稳定度(M值),M>1.5的内参基因不可用,M越小,内参基因的表达越稳定。该软件分析结果显示:6个候选内参基因的M值均<1.5,PRS27a和RPS3的表达最稳定,其后依次是α-tubulin、RPL13α和β-actin,GAPDH的表达最不稳定。NormFinder通过比较样本的组内变异和组间变异来评估基因的稳定值。该软件分析结果显示:RPL13α的表达最稳定,其后依次是α-tubulin、RPS3、RPS27a和β-actin,GAPDH的表达最不稳定。BestKeeper是以标准差和P来判断内参基因是否可用。该软件分析结果显示:所有候选内参基因的标准差均<1,但β-actin和GAPDH P>0.05,不适合作为本试验条件下的内参,其余4个内参基因可用。综合3个软件的评价结果发现:RPS3、PRS27a、RPL13α和α-tubulin这4个内参的表达稳定性排名稍有差异,RPS3的表达最稳定,PRS27a和RPL13α次之,随后是α-tubulin(表2)。GeNorm软件通过判断配对变异值(<0.15)来确定内参基因最佳数目。由表3可知:V2/3=0.084<0.15,表明该条件下最佳内参基因的数目n=2(表3)。
表 2 利用BestKeeper软件评价候选内参基因的表达稳定性
Table 2. Stability of candidate reference genes based on BestKeeper analysis
基因 GeNorm NormFinder BestKeeper 稳定值 排名 稳定值 排名 标准差 P 排名 β-actin 0.391 4 0.244 5 0.27 0.313 6 GAPDH 0.426 5 0.293 6 0.16 0.261 5 α-tubulin 0.242 2 0.177 2 0.24 0.042 4 RPL13α 0.319 3 0.164 1 0.32 0.024 3 RPS27a 0.197 1 0.206 4 0.35 0.012 2 RPS3 0.197 1 0.202 3 0.39 0.002 1 表 3 内参基因的最佳数量评估
Table 3. Determination of the optimal number of reference genes for normalization
Vn/n+1 配对变异值 Vn/n+1 配对变异值 V2/3 0.084 V4/5 0.092 V3/4 0.092 V5/6 0.074 说明:n表示内参基因数目 -
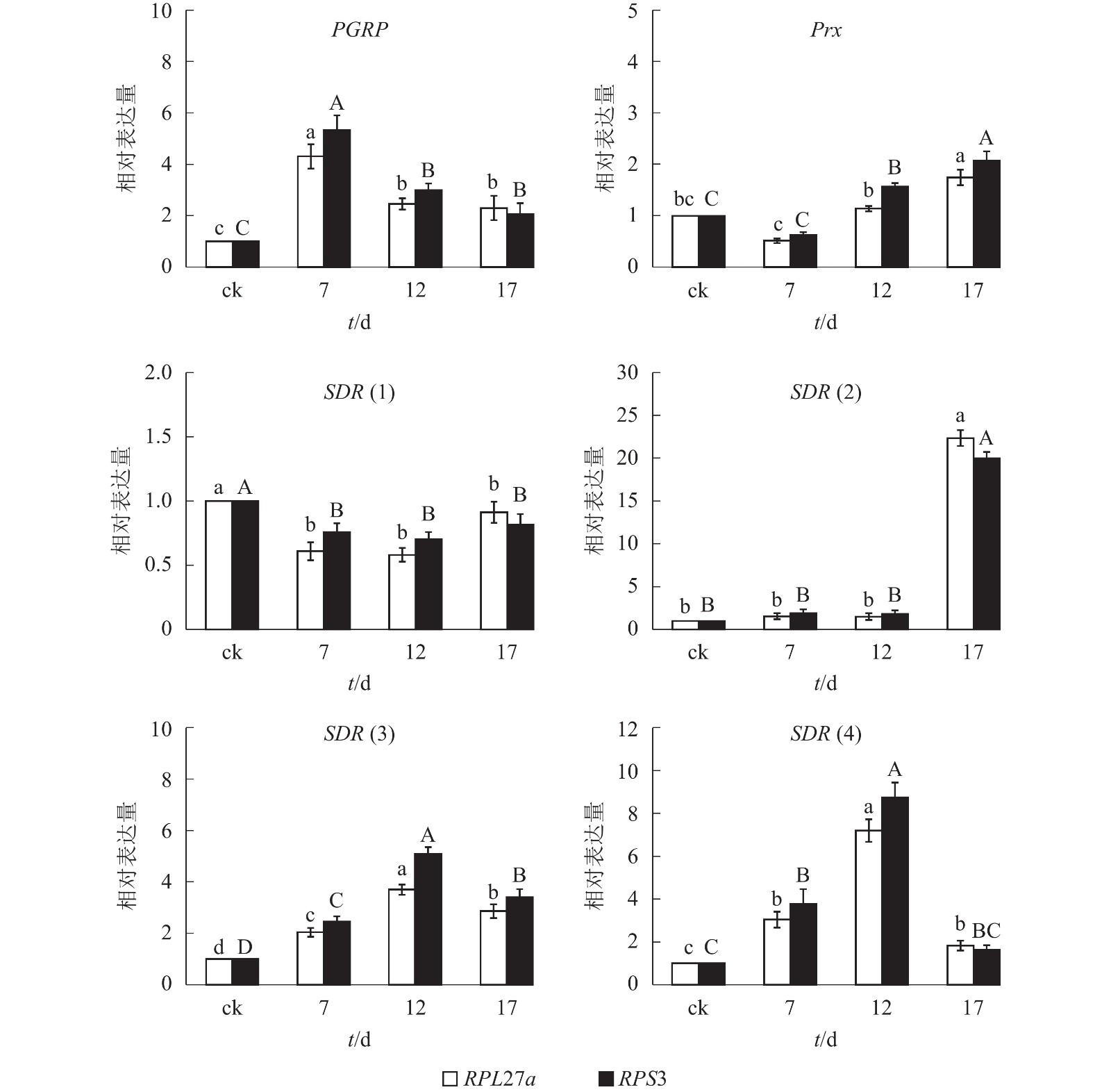
综合3款软件的内参基因表达稳定性排名和最佳组合数目分析结果,本研究选择PRS27a和RPS3为内参基因。由图3可知:以PRS27a和RPS3为内参基因时,6个目的基因在平沙绿僵菌不同处理时间(0、7、12和17 d)的筛胸梳爪叩甲幼虫中的表达变化趋势相同;其中,PGRP基因相对表达量在7 d时都较对照(0 d)显著提高(P<0.05),随后下降,12 和17 d时无显著差异;Prx基因相对表达量整体呈上升趋势,在12 和17 d时均显著高于对照(P<0.05);SDR(1)相对表达量则呈下降趋势;SDR(2)在17 d 时相对表达量显著升高(P<0.05);SDR(3)和SDR(4)相对表达量均先升高后下降,并在12 d时表达量最高,较其他处理时间存在显著性差异(P<0.05)。
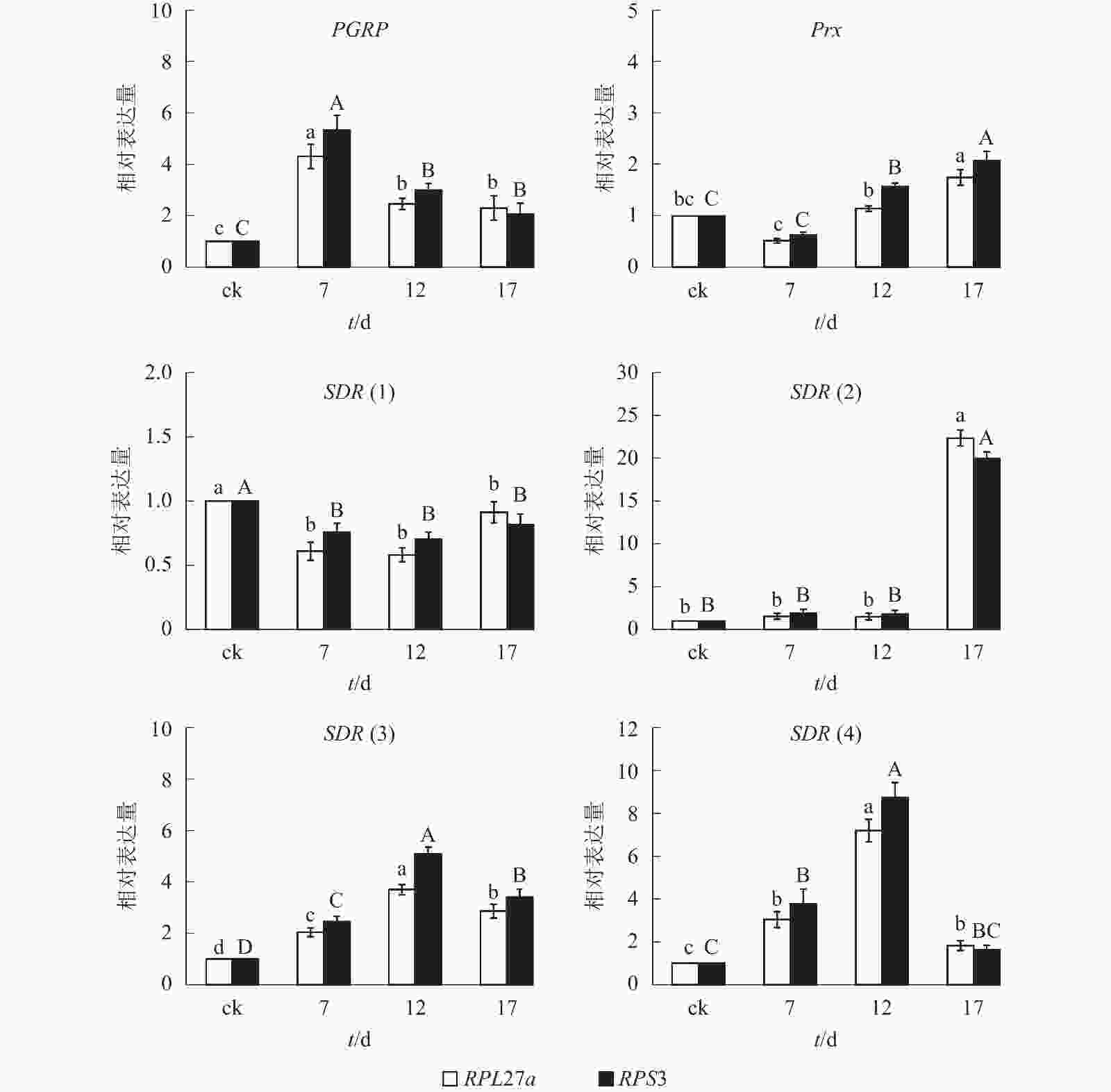
图 3 以PRS27a和RPS3为内参时6个目的基因不同处理时间下的相对表达量
Figure 3. Expression analysis of six target genes under different processing times calibrated by PRS27a and RPS3 as reference genes
-
实时荧光定量PCR具有快速、准确和灵敏的特性,在基因表达分析研究中应用广泛,然而表达稳定的内参基因选择是获得目的基因准确表达结果的前提条件。同一内参基因在不同物种、组织、发育阶段和处理条件下的表达稳定性也存在差异[8-9]。目前尚未见有关筛胸梳爪叩甲幼虫内参基因的研究报道。本研究首次对该虫在平沙绿僵菌侵染条件下的候选内参基因进行了筛选评估。GeNorm、NormFinder和BestKeeper软件对候选内参基因的分析结果表明:PRS27a和RPS3的表达稳定。本研究以PGRP、Prx、SDR(1)、SDR(2)、SDR(3)和 SDR(4)为目的基因对PRS27a和RPS3内参基因进行了验证,结果显示:分别以这2个基因为内参时,6个目的基因的表达趋势一致,证明了这2个基因作为内参的可靠性。PGRP隶属于模式识别受体蛋白(pattern recognition receptor proteins,PRRs)家族,可与革兰氏阳性菌和革兰氏阴性菌的肽聚糖结合,激活Toll和Imd信号转导通路,诱导免疫防御相关的水解级联反应,发生吞噬作用[22-23]。以PRS27a和RPS3为内参时,目的基因PGRP在筛胸梳爪叩甲幼虫体内被平沙绿僵菌诱导表达,7 d时较对照组显著升高,该现象与赤拟谷盗和油菜花露尾甲Meligethes aeneus PGRP基因被寄生物或病原菌诱导后表达上调相似[24-25]。Prx基因编码过氧化物还原酶(peroxiredoxin),是抗氧化家族中的一员,能够通过消除细胞内的活性氧(ROS)以维持氧化还原平衡[26]。以PRS27a和RPS3为内参时,目的基因Prx整体表达呈上升趋势,推测其可能在病程后期发挥作用。SDR蛋白(short-chain dehydrogenases/reductases,短链脱氢还原酶)是一个超级家族,由多种酶编码基因家族组成,在多种生物合成途径、细胞信号通路和病理途径中都扮演重要角色[27]。本研究发现:筛胸梳爪叩甲幼虫4个SDR基因[SDR(1)、SDR(2)、SDR(3)和 SDR(4)]对绿僵菌侵染的响应机制不同,可能是因为参与过程不同。由上可见,以PRS27a和RPS3为内参时,筛胸梳爪叩甲幼虫PGRP、Prx和SDR这3类基因的表达模式符合赤拟谷盗和油菜花露尾甲等昆虫对病原菌的响应机制,进一步证实了该内参组合适用于绿僵菌侵染条件下的目的基因表达水平研究。
本研究首次报道了在平沙绿僵菌侵染条件下筛胸梳爪叩甲幼虫可用的内参基因(PRS27a和RPS3),为后续研究绿僵菌和竹林金针虫互作相关基因的表达奠定基础,也对其他昆虫基因表达以及虫菌互作相关研究提供参考。
Screening and application of reference genes for qRT-PCR in bamboo wireworm
-
摘要:
目的 筛选平沙绿僵菌Metarhizium pingshaense侵染条件下筛胸梳爪叩甲Melanotus cribricollis幼虫稳定表达的内参基因,为该竹林金针虫相关基因的表达研究奠定基础。 方法 基于筛胸梳爪叩甲幼虫的转录组数据,利用实时荧光定量PCR扩增特异性引物,分析相关性和扩增效率;利用GeNorm、NormFinder和BestKeeper软件评估筛选出6个候选内参基因(β-actin、GAPDH、α-tubulin、RPL13α、RPS3、RPS27a),并验证其表达稳定性。 结果 GeNorm分析结果显示:PRS27a和RPS3的表达最稳定,随后依次是α-tubulin、RPL13α、β-actin和GAPDH;最适合的内参基因数目为2。NormFinder分析结果显示:RPL13α的表达最稳定,随后依次是α-tubulin、RPS3、RPS27a、β-actin和GAPDH。BestKeeper分析结果显示:β-actin和GAPDH的P>0.5,不适合作为本试验条件下的内参基因。不同软件分析得出的候选内参排序存在一定差异。综合分析和表达稳定性验证表明PRS27a或RPS3是最佳内参基因,6个目的基因的表达水平变化趋势均基本一致。 结论 PRS27a和RPS3是研究平沙绿僵菌侵染的竹林金针虫相关基因表达的最佳内参基因。图3表3参27 Abstract:Objective The objective of this study was to screen the stable expression of internal reference genes of Melanotus cribricollis larvae infected by Metarhizium pingshaense, so as to lay a foundation for the research on related gene expression in this bamboo wireworm. Method Based on the transcriptome data of M. cribricollis larvae, the correlation (R2) and amplification efficiency were analyzed by qRT-PCR with specific primers. The 6 candidate reference genes including β-actin, GAPDH, α-tubulin, RPL13α, RPS3 and RPS27a were evaluated by GeNorm, NormFinder and BestKeeper softwares. The stabilities of selected candidate reference genes including PRS27a and RPS3 were further validated by analyzing the expression of 6 target genes. Result GeNorm analysis showed that the expression of PRS27a and RPS3 were the most stable, followed by α-tubulin, RPL13α, β-actin and GAPDH. The most suitable number of internal reference genes was 2. NormFinder analysis showed that the expression of RPL13α was the most stable, followed by α-tubulin, RPS3, RPS27a, β-actin, and GAPDH. BestKeeper analysis showed that the P values of β-actin and GAPDH were >0.5, which were not suitable for reference genes under the condition of this experiment. There were some differences in the ranking of candidate internal parameters obtained by different software analysis. Comprehensive analysis and expression stability verification showed that PRS27a or RPS3 were the best internal reference genes, and the expression levels of 6 target genes were basically the same. Conclusion PRS27a and RPS3 were the most appropriate reference genes for qRT-PCR analysis in bamboo wireworm infected by M. pingshaense. [Ch, 3 fig. 3 tab. 27 ref.] -
图 2 筛胸梳爪叩甲幼虫候选内参基因的Ct值分析
Figure 2 Ct value analysis of candidate reference genes of Melanotus cribricollis larvae
图 3 以PRS27a和RPS3为内参时6个目的基因不同处理时间下的相对表达量
对照组的相对表达量设定为1 。不同小写字母表示以RPL27a为内参时差异显著(P<0.05),不同大写字母表示以PRS3为内参时差异显著(P<0.05)
Figure 3 Expression analysis of six target genes under different processing times calibrated by PRS27a and RPS3 as reference genes
表 1 qRT-PCR引物信息
Table 1. Informations of qRT-PCR primers
基因 引物序列 产物长度/bp 扩增效率/% R2 β-actin F:GGATACCTCTTTTGCTCTGGG,R:ATCAGGGTGTCATGGTTGG 75 112.48 0.993 GAPDH F:CTACTCATGGTCGTTACAAGGG,R:TTCTACAACGTATTCAGCTCCAG 140 101.79 0.993 α-tubulin F:GAAGCTCGTGAAGATTTGGC,R:ACCTTCGCCTTCTCCTTCTC 137 100.63 0.996 RPL13α F:CTGAGGAAGAGCGTAAGGTG,R:TCAGCACGAGCCTTTCTTAAG 145 106.97 0.990 RPS27a F:CTTGTCCTGAATCTTTGCCTTG,R:GTTCTTTTGGTAGCGTGTCATG 146 96.20 0.998 RPS3 F:CAATAGCGCACAAACCACG,R:TGTATTGGGTGAAAAGGGAAGG 128 111.93 0.992 PGRP F:TGTCGTACTTCTGGCTATCATTG,R:TGTGATGGAGGGTTTACTTGC 123 88.69 0.989 Prx F:CTATCCCTTAGACTTCACCTTCG,R:ATTTCTCCCAAACCTCCCTG 171 106.67 0.996 SDR(1) F:CGGCATTGACGGAAACTTTAC,R:GGTTTCCACAGACTTTTGCG 126 109.16 0.995 SDR(2) F:AGGTGCTAGTTCGGGAATTG,R:CGTGTAATTTGCCAGGTTTTCC 140 99.49 0.997 SDR(3) F:GGATTACGAGCATAAGTCCTGG,R:CGGCGATGTCTTCAGATTTTAAC 126 106.58 0.996 SDR(4) F:TTAGGGTTTCAGTCAAGGCAG,R:GAAGCCGTCCAAGATATGAAAG 148 93.34 0.994  下载: 导出CSV
下载: 导出CSV
表 2 利用BestKeeper软件评价候选内参基因的表达稳定性
Table 2. Stability of candidate reference genes based on BestKeeper analysis
基因 GeNorm NormFinder BestKeeper 稳定值 排名 稳定值 排名 标准差 P 排名 β-actin 0.391 4 0.244 5 0.27 0.313 6 GAPDH 0.426 5 0.293 6 0.16 0.261 5 α-tubulin 0.242 2 0.177 2 0.24 0.042 4 RPL13α 0.319 3 0.164 1 0.32 0.024 3 RPS27a 0.197 1 0.206 4 0.35 0.012 2 RPS3 0.197 1 0.202 3 0.39 0.002 1
下载: 导出CSV
表 3 内参基因的最佳数量评估
Table 3. Determination of the optimal number of reference genes for normalization
Vn/n+1 配对变异值 Vn/n+1 配对变异值 V2/3 0.084 V4/5 0.092 V3/4 0.092 V5/6 0.074 说明:n表示内参基因数目
下载: 导出CSV
-
[1] 邓顺, 舒金平, 王浩杰. 筛胸梳爪叩甲幼虫寄主调查及其土壤空间分布[J]. 昆虫知识, 2010, 47(5): 983 − 987. DENG Shun, SHU Jinping, WANG Haojie. Investigation of host rang of wireworms (Melanotus cribricollis) and their spatial distribution in soil [J]. Chin Bull Entomol, 2010, 47(5): 983 − 987. [2] 舒金平, 滕莹, 陈文强, 等. 筛胸梳爪叩甲的防治技术研究[J]. 林业科学研究, 2012, 25(5): 620 − 625. SHU Jinping, TENG Ying, CHEN Wenqiang, et al. Control techniques of Melanotus cribricollis (Coleoptera: Elateridae) [J]. For Res, 2012, 25(5): 620 − 625. [3] SCHOLTE E J, KNOLS B G J, TAKKEN W. Infection of the malaria mosquito Anopheles gambiae with the entomopathogenic fungus Metarhizium anisopliae reduces blood feeding and fecundity [J]. J Invertebr Pathol, 2006, 91(1): 43 − 49. [4] HOWARD A F V, N’GUESSAN R, KOENRAADT C J M, et al. The entomopathogenic fungus Beauveria bassiana reduces instantaneous blood feeding in wild multi-insecticide-resistant Culex quinquefasciatus mosquitoes in Benin, West Africa [J]. Parasites Vectors, 2010, 3(1): 87. doi: 10.1186/1756-3305-3-87. [5] ANSARI M A, BUTT T M. Influence of the application methods and doses on the susceptibility of black vine weevil larvae Otiorhynchus sulcatus to Metarhizium anisopliae in field-grown strawberries [J]. BioControl, 2013, 58(2): 257 − 267. [6] IBRAHIM A A, MOHAMED H F, EL-NAGGAR S E M, et al. Isolation and selection of entomopathogenic fungi as biocontrol agent against the greater wax moth, Galleria mellonella L. (Lepidoptera: Pyralidae) [J]. Egypt J Biol Pest Control, 2016, 26(2): 249 − 253. [7] DERVEAUX S, VANDESOMPELE J, HELLEMANS J. How to do successful gene expression analysis using real-time PCR [J]. Methods, 2010, 50(4): 227 − 230. [8] de JONGE H J M, FEHRMANN R S N, de BONT E S J M, et al. Evidence based selection of housekeeping genes [J]. PLoS One, 2007, 2(9): e898. doi: 10.1371/journal.pone.0000898. [9] KOZERA B, RAPACZ M. Reference genes in real-time PCR [J]. J Appl Genet, 2013, 54(4): 391 − 406. [10] 符伟, 谢文, 张卓, 等. Bt 毒素诱导下小菜蛾实时定量PCR内参基因的筛选[J]. 昆虫学报, 2012, 55(12): 1406 − 1412. FU Wei, XIE Wen, ZHANG Zhuo, et al. Selection of valid reference genes for gene expression studies by quantitative real-time PCR in Plutella xylostella (Lepidotera: Plutellidae) after exposure to Bt toxin [J]. Acta Entomol Sin, 2012, 55(12): 1406 − 1412. [11] 冯波, 郭前爽, 毛必鹏, 等. 松墨天牛化学感受组织荧光定量PCR内参基因的鉴定与筛选[J]. 昆虫学报, 2016, 59(4): 427 − 437. FENG Bo, GUO Qianshuang, MAO Bipeng, et al. Indetification and selection of valid reference genes for assaying gene expression in the chemosensory tissues of Monochamus alternates (Coleoptera: Cerambycidae) by RT-qPCR [J]. Acta Entomol Sin, 2016, 59(4): 427 − 437. [12] 刘金泊, 欧静, 姚富姣, 等. 磷化氢诱导下赤拟谷盗实时定量PCR内参基因的筛选[J]. 农业生物技术学报, 2014, 22(2): 257 − 264. LIU Jinbo, OU Jing, YAO Fujiao, et al. Identification of appropriate reference genes for gene expression studies by quantitative real-time PCR in Tribolium castaneum after exposure to phosphine [J]. J Agric Boitechnol, 2014, 22(2): 257 − 264. [13] 杨苓, 胡晓静, 徐志峰, 等. 桃蛀螟实时荧光定量PCR内参基因的筛选[J]. 昆虫学报, 2017, 60(11): 1266 − 1277. YANG Ling, HU Xiaojing, XU Zhifeng, et al. Screening of reference genes for qRT-PCR in Conogethes punctiferails (Lepidotera: Crambidae) [J]. Acta Entomol Sin, 2017, 60(11): 1266 − 1277. [14] 陶蓉, 李慧, 孙宇航, 等. 美国白蛾内参基因的鉴定及筛选[J]. 林业科学, 2019, 55(9): 111 − 120. TAO Rong, LI Hui, SUN Yuhang, et al. Indetification and screening of internal reference genes of Hyphantria cunea (Lepidotera: Arctiidae) [J]. Sci Silv Sin, 2019, 55(9): 111 − 120. [15] 张亚波, 吴盼盼, 王鹏, 等. 一株绿僵菌的鉴定及其生物学特性[J]. 林业科学, 2012, 48(12): 134 − 140. ZHANG Yabo, WU Panpan, WANG Peng, et al. Identification and biological characteristics of a Metarhizium pingshaense strain isolated from Melanotus cribricollis larva [J]. Sci Silv Sin, 2012, 48(12): 134 − 140. [16] YE Bihuan, ZHANG Yabo, SHU Jinping, et al. RNA-sequencing analysis of fungi-induced transcripts from the bamboo wireworm Melanotus cribricollis (Coleoptera: Elateridae) larvae [J]. PLoS One, 2018, 13(1): e0191187. doi: 10.1371/journal.pone.0191187. [17] RADONIĆ A, THULKE S, MACKAY I M, et al. Guideline to reference gene selection for quantitative real-time PCR [J]. Biochem Biophys Res Commun, 2004, 313(4): 856 − 862. [18] VANDESOMPELE J, de PRETER K, PATTYN F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes [J]. Genome Biol, 2002, 3(7): 1 − 11. [19] ANDERSEN C L, JENSEN J L, ØRNTOFT T F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets [J]. Cancer Res, 2004, 64(15): 5245 − 5250. [20] PFAFFL M W, TICHOPAD A, PRGOMET C, et al. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: bestkeeper-Excel-based tool using pair-wise correlations [J]. Biotechnol Lett, 2004, 26: 509 − 515. [21] 吴建阳, 何冰, 杜玉洁, 等. 利用geNorm、NormFinder和BestKeeper软件进行内参基因稳定性分析的方法[J]. 现代农业科技, 2017(5): 278 − 281. WU Jianyang, HE Bing, DU Yujie, et al. Analysis method of systematically evaluating stability of reference genes using geNorm, NormFinder and BestKeeper [J]. Modern Agric Sci Technol, 2017(5): 278 − 281. [22] LEMAITRE B, HOFFMANN J. The host defense of Drosophila melanogaster [J]. Annu Rev Immunol, 2007, 25(1): 697 − 743. [23] VOGEL H, BADAPANDA C, VILCINSKAS A. Identification of immunity-related genes in the burying beetle Nicrophorus vespilloides by suppression subtractive hybridization [J]. Insect Mol Biol, 2011, 20(6): 787 − 800. [24] ZHONG Daibin, WANG Meihui, PAI A, et al. Transcription profiling of immune genes during parasite infection in susceptible and resistant strains of the flour beetles (Tribolium castaneum) [J]. Exp Parasitol, 2013, 134(1): 61 − 67. [25] VOGEL H, BADAPANDA C, KNORR E, et al. RNA sequencing analysis reveals abundant developmental stage-specific and immunity- related genes in the pollen beetle Meligethes aeneus [J]. Insect Mol Biol, 2014, 23(1): 98 − 112. [26] RHEE S G, KANG S W, CHANG T S. Peroxiredoxin, a novel family of peroxidases [J]. IUBMB Life, 2001, 52(1/2): 35 − 41. [27] KAVANAGH K L, JÖRNVALL H, PERSSON B, et al. The SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes [J]. Cell Mol Life Sci, 2008, 65(24): 3895 − 3906. -
-
链接本文:
https://zlxb.zafu.edu.cn/article/doi/10.11833/j.issn.2095-0756.20200492
 点击查看大图
点击查看大图

计量
- 文章访问数: 3351
- HTML全文浏览量: 984
- PDF下载量: 41
- 被引次数: 0

