





-
毛竹Phyllostachys edulis是中国亚热带地区分布最广泛的竹种,约占中国竹林种植面积的73.76%,具有极高的生态价值和经济价值。但在非生物胁迫下,毛竹林的种植面积显著减少[1]。毛竹在生长发育过程中经历多种胁迫后会引发一系列生理生化变化,最终影响毛竹的正常生长[2]。
毛竹基因组由超过63.24%的转座子(TE)组成[3]。TE可以通过2种主要机制在基因组内移动:复制-粘贴和剪切-粘贴。反转录转座子(Ⅰ类)通过RNA中间体利用复制-粘贴机制,而DNA转座子(Ⅱ类)直接作为DNA片段利用剪切-粘贴或复制-粘贴机制在基因组内移动[4−5]。TE是真核生物基因组的动态组成部分,可以被激活或沉默。TE受表观遗传变化的影响较大[6−7],可以通过塑造表观基因组促进植物对非生物胁迫的适应性反应。对水稻Oryza sativa研究表明:高盐和低温胁迫可诱导基因转录起始位点100 bp内插入TE[8]。转座子受逆境胁迫更容易发生转录[9]。大麦Hordeum vulgare基因组中的BARE-1反转录转座子具有转录活性,拷贝数与基因组大小、温度、水分、土壤类型、海拔等呈正相关[10]。
不确定编码潜力转录本(TUCP)[11]是一类具有明显组织特异性,与基因间长链非编码RNA表达模式相似,但序列保守性明显更高的一种转录本[12]。TUCP可以作为非编码调节因子,翻译成小肽或在转录调节、细胞分化等多种生物活动中发挥作用[13]。越来越多的真核生物基因组TUCP被高通量测序鉴定出来,如棉花Gossypium spp.[14]、人类Homo sapiens [15]、罗非鱼Oreochromis mossambicus[16]、山羊Capra hircus[17]和杜仲Eucommia ulmoides[18]等。在拟南芥Arabidopsis thaliana、水稻和玉米Zea mays中,TE衍生的TUCP参与低温和盐的胁迫反应,并影响种子发芽和幼苗绿化率[19]。棉花中TUCP参与光合作用并影响生长发育过程[14]。高温胁迫影响TUCP转录从而使红枣Ziziphus jujuba基因表达产生了整体性改变[20]。
目前,对毛竹转座子衍生TUCP (TE-TUCP)表达的综合分析尚未见报道。鉴于此,本研究调查非生物胁迫下毛竹幼苗TE-TUCP活性的变化及附近基因的差异表达情况,以期为转座子参与毛竹抗逆分子机制提供参考。
-
采用单株母竹的毛竹种子进行水培育苗,每盆1株毛竹幼苗,每组3盆,培养至毛竹幼苗五叶一心期。处理过程如下:首先,在25 ℃暗培养箱中培养幼苗3 d,以避免可见光下紫外线的影响。接着,对第1组幼苗进行冷胁迫处理(C4),即在4 ℃下处理16 h;对第2组幼苗进行热应激处理(H42),即在42 ℃下处理4 h;对第3组幼苗进行紫外线胁迫处理(UV),在30 W紫外线下处理2 h;第4组幼苗未经以上任何胁迫处理,作为对照组(ck);对第5组幼苗进行盐胁迫处理(Sa),水培培养基中配置浓度为200 mmol·L−1氯化钠溶液培育幼苗3 d;对第6组幼苗多次浇水,保持与第5组等体积的水培培养基,培育幼苗3 d,作为盐胁迫处理的对照组(wa)。所有样品均在当天10:00采集后在液氮中冷冻保存。
-
利用DING等[21]对毛竹幼苗4个胁迫处理(低温、高温、高盐、紫外照射)的全转录组数据(登录号:PRJNA826540),分析和鉴定基因表达水平。
用Trimmomatic[22]软件进行质量控制;使用Hisat 2[23]软件将质控后获得的干净数据(clean reads)与毛竹参考基因组[3]进行序列比对获得bam类型文件;利用featureCounts软件[24]计数,得到每个基因在各个样本中的原始计数(raw counts,RC)。将原始计数导入R中,利用DESeq 2[25]进行差异表达分析。在读取计数值后,对表达值进行归一化,并计算差异倍数(FC)。根据log2|FC|>0和P<0.05选择差异表达基因。
-
利用相同的全转录组测序数据,分析和鉴定TUCP表达水平。具体步骤如下:①数据预处理。首先使用Cuffmerge软件[26],对各样品拼接得到的转录本进行合并,去掉其中链方向不确定的转录本,得到本次测序完整的转录组信息。之后,对合并的转录本集合进行TE-TUCP的筛选。②转录本外显子(exon)个数筛选。过滤转录本拼接结果中大量低表达量、低可信度的单外显子转录本,选择exon≥2个的转录本。③转录本长度筛选。选择长度>200 bp的转录本。④mRNA剔除。通过Cuffcompare软件,筛除具有编码能力的mRNA,将余下的拼接转录本中,与毛竹基因组编码基因exon区域有重叠的转录本作为后续分析的RNA。⑤转录本表达量筛选。通过Cuffquant计算每条转录本的表达量,选择RC≥1的转录本。⑥针对拼接得到的转录本,剔除mRNA转录本后,综合CNCI[27]、PFAM[28]、CPC2[29]等编码潜能分析方法,进行RNA编码能力分析,将3个软件均表明无编码能力的转录本定义为lncRNA (long non-coding RNA);其余有部分编码能力,且至少包含1个开放阅读框(ORF)且非lncRNA转录本的即为TUCP。
-
利用ZHOU等[30]鉴定的毛竹基因组转座子,使用RepeatMasker[31]鉴定毛竹参考基因组[3]中的转座子,分析已鉴定的转座子在毛竹基因组中的分布和含量。
-
使用bedtools [32]分析TE和TUCP关系,以确定TE是否与TUCP重叠。与TE序列重叠超过60%的TUCP被鉴定为TE衍生的,简称为TE-TUCP。为了评估TE-TUCP的表达模式,使用StringTie[33]的合并函数组装TE转录本,合并至少1 bp的重叠区域,直到没有交集。随后,使用Gffcompare评估组装TE转录本的质量[34],最后估计TE-TUCP的表达丰度。将组装后的数据利用featureCounts软件对bam类型的输入文件进行计数,得到表达矩阵。使用tximport R包[35]将featureCounts输出的表达矩阵转换为DESeq2所需的格式。然后使用DESeq2[25]识别差异表达的TE-TUCP。P<0.05且log2|FC|>0为显著差异表达的TE-TUCP。将能转录出至少1个TE-TUCP的转座子定义为TUCP-TE;否则,定义为nonTUCP-TE。
-
使用perl脚本check_GCbG_PlusForTE.pl程序分别筛选TUCP-TE和nonTUCP-TE附近5 000 bp差异表达基因。使用OmicShare (https://www.omicshare.com/tools/)上的Gogsea对差异表达基因进行基因本体(GO)富集分析。组间样本基因表达差异筛选标准为:log2|FC|>0且P<0.05。保留前30条GO条目,通过矩阵点图显示结果。
-
为了验证转录组测序结果的准确性,使用天根生化科技有限公司的RNA提取试剂盒(DP441)提取总RNA。利用TaKaRa Bio公司的反转录试剂盒(RR047A)合成cDNA,用RT-qPCR检测相对转录水平。从鉴定的差异表达TE-TUCP中随机挑选4个TE-TUCP及其附近基因,利用PrimerPremier 6设计RT-qPCR引物,使用翌圣生物科技股份有限公司的Hieff® qPCR SYBR Green Master Mix (No Rox)(11201ES)进行RT-qPCR试验,反应体系和程序参照试剂说明书,以毛竹肌动蛋白(Actin)作为内参基因。每个RT-qPCR设置3个生物重复和3个技术重复。采用2−∆∆Ct计算相对基因表达水平[36]。使用Graphpad可视化绘图。
-
毛竹幼苗全转录组测序数据共获得1 749 524个转录本。在这些转录本的基础上使用Cuffmerge进行数据预处理,保留1 008 312个转录本。通过表达量筛选,获得567 404个表达量至少为1的转录本。
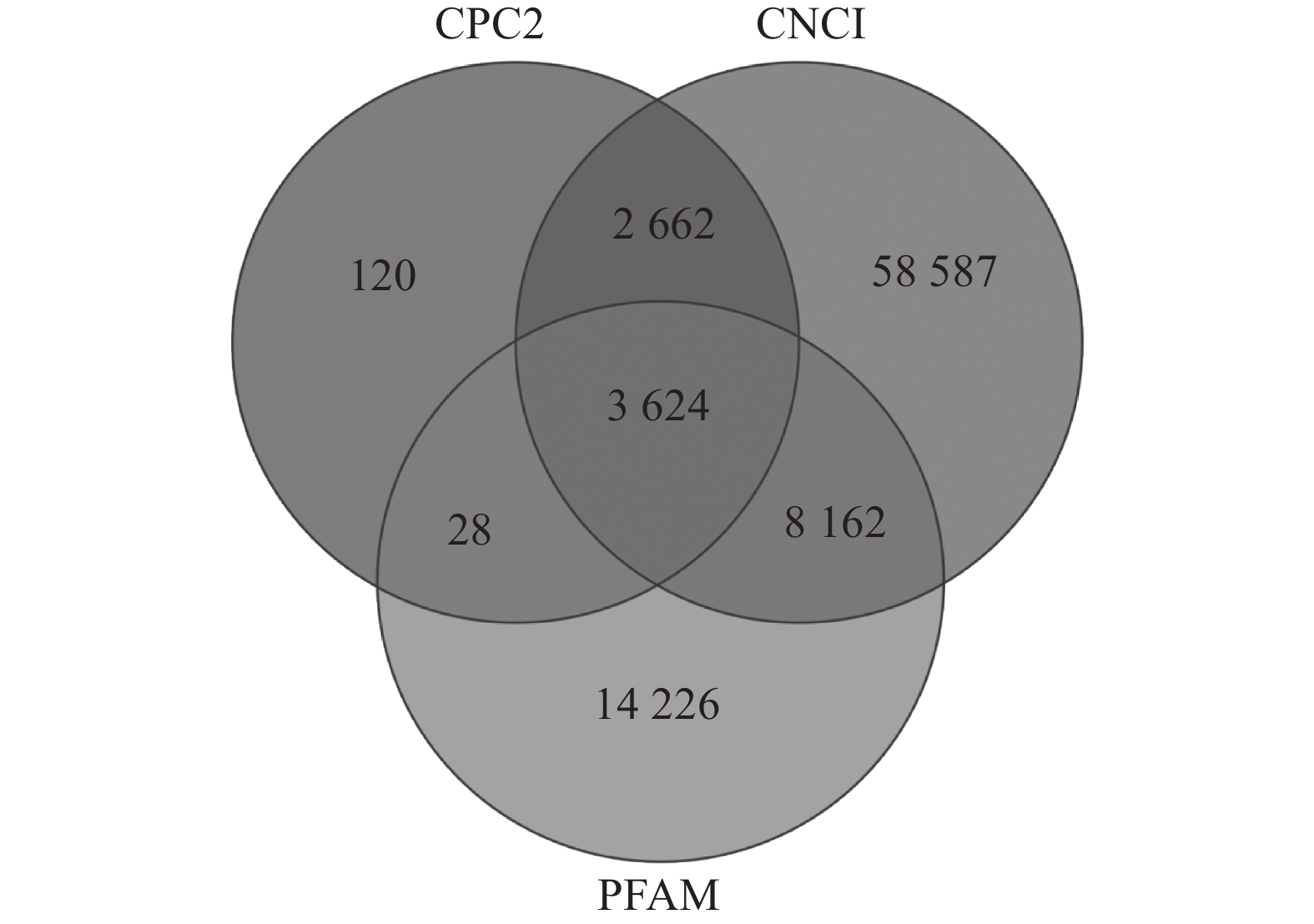
如图1所示:CPC2预测出6 434个TUCP,CNCI预测出73 035个TUCP,PFAM预测出26 040个TUCP,共预测出87 409个TUCP。对TUCP的结构分析表明:TUCP的平均长度为759.73 bp,比mRNA的平均长度短,但比lncRNA的平均长度(241.02 bp)长。
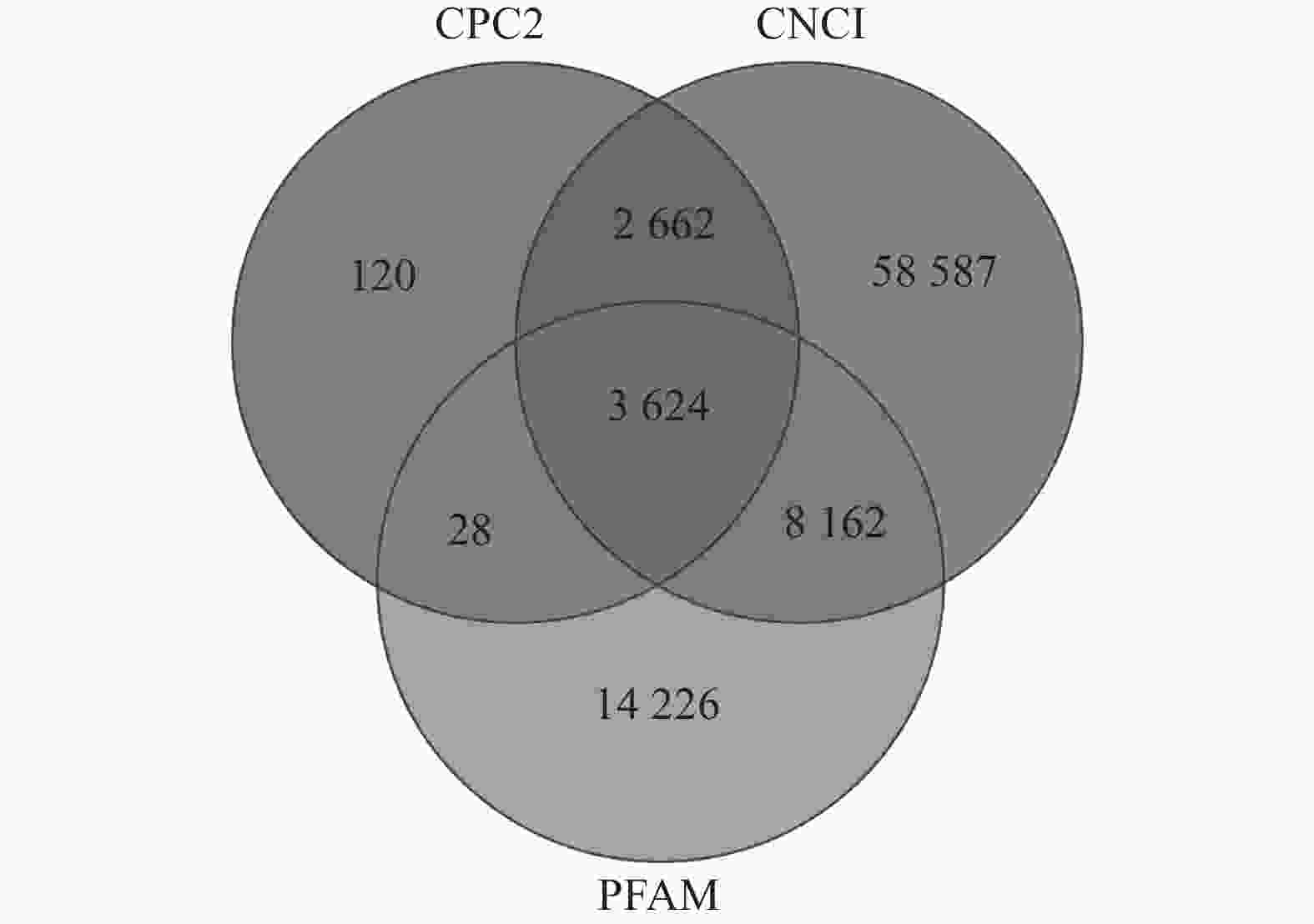
图 1 TUCP筛选结果
Figure 1. Results of TUCP screening
-
通过RepeatMasker对毛竹参考基因组注释,共得到991 865条转座子。根据上述鉴定的87 409个TUCP在基因组的位置和转座子在基因组的位置,共有57 627个TUCP与TE序列重叠超过60%,命名为TE-TUCP。其中来源反转录转座子(RE)的TE-TUCP非常丰富,占73.60%。
如表1所示:鉴定的转座子中,TE-TUCP中有17 265个来源于LTR/Copia超家族,23 980个来源于LTR/Gypsy超家族。LTR/Copia和LTR/Gypsy来源的TUCP数量占所有TE-TUCP的71.57%,说明LTR-TE-TUCP含量丰富,可能与LTR/Copia和LTR/Gypsy在毛竹基因组含量丰富(54.97%)[37]有关。DNA-TE-TUCP占所有转座子来源的TUCP的20.75%。其中DNA/CMC-EnSpm类型转座子(66.39%)和DNA/MULE-MuDR类型转座子(55.56%)在TUCP-TE中增加,可能是非生物胁迫导致。
表 1 毛竹TE-TUCP转座子来源
Table 1. Sources of TE-TUCP transposons in Ph. edulis
转座子来源分类 TUCP-TE/
个nonTUCP-TE/
个数量合
计/个DNA 45 115 160 DNA/CMC-EnSpm 2 994 1 516 4 510 DNA/hAT 4 18 22 DNA/hAT-Ac 745 907 1 652 DNA/hAT-Tag1 17 26 43 DNA/hAT-Tip100 228 209 437 DNA/MULE-MuDR 2 368 1 894 4 262 DNA/PIF-Harbinger 112 154 266 DNA/TcMar-Stowaway 153 452 605 LINE/L1 1 040 1 050 2 090 Low_complexity 4 9 13 LTR 68 1058 1126 LTR/Caulimovirus 6 39 45 LTR/Copia 8 456 8 809 17 265 LTR/Gypsy 10 662 13 318 23 980 Other/centromeric 2 2 4 RC/Helitron 241 427 668 Simple_repeat 112 341 453 SINE/L1 6 6 12 SINE 2 2 未知 12 12 合计 27 263 30 364 57 627 -
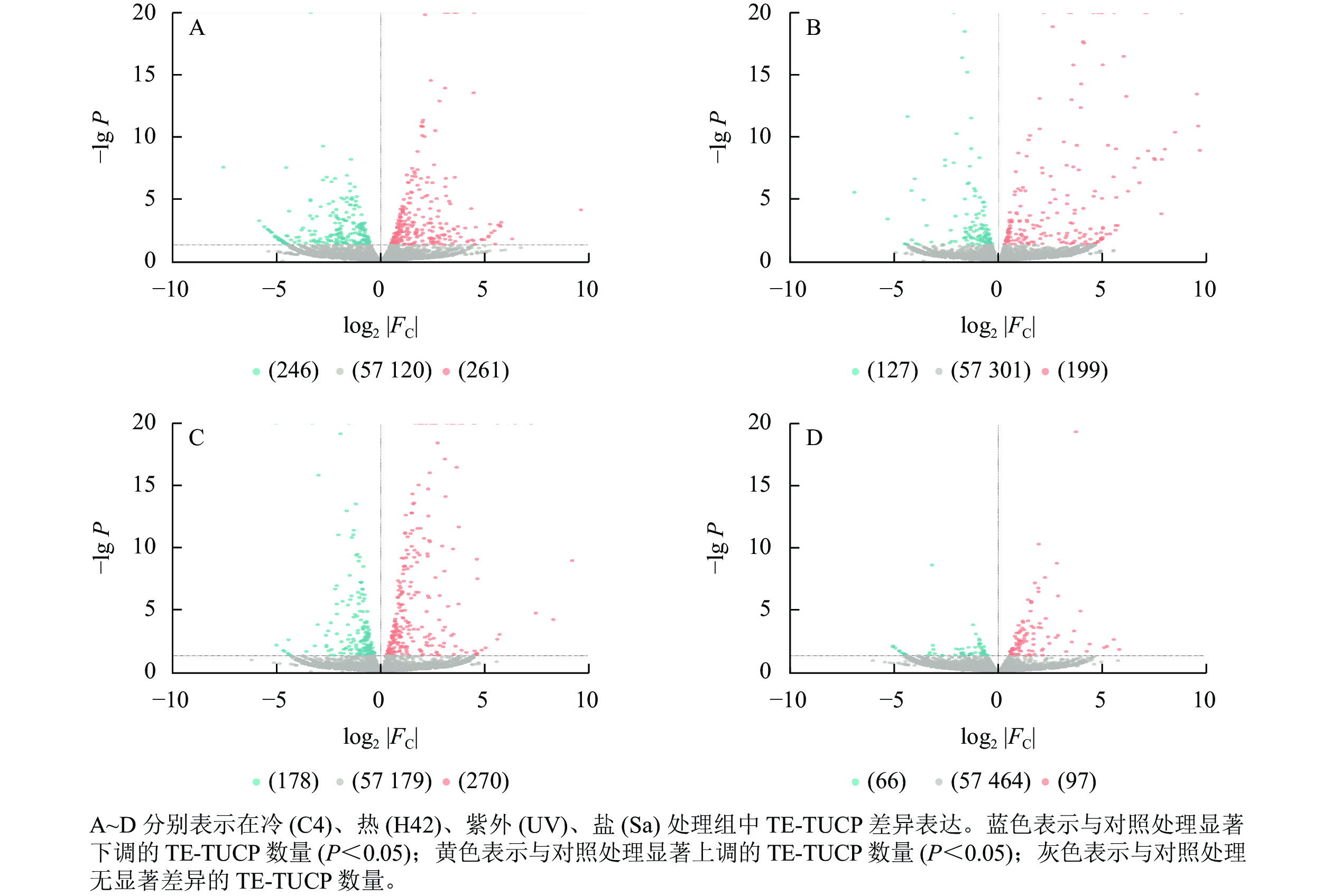
从图2可见:C4与ck处理组共筛选出261个上调TE-TUCP,246个下调TE-TUCP (图2A)。H42与ck处理组共筛选出199个上调TE-TUCP,127个下调TE-TUCP (图2B)。UV与ck处理组共筛选出270个上调TE-TUCP,178个下调TE-TUCP(图2C)。Sa与wa处理组共筛选出97个上调TE-TUCP,66个下调TE-TUCP (图2D)。TE-TUCP表现出胁迫特异性表达模式,在冷、盐胁迫下下调个数多于上调个数,在热和紫外胁迫下上调个数多于下调个数。
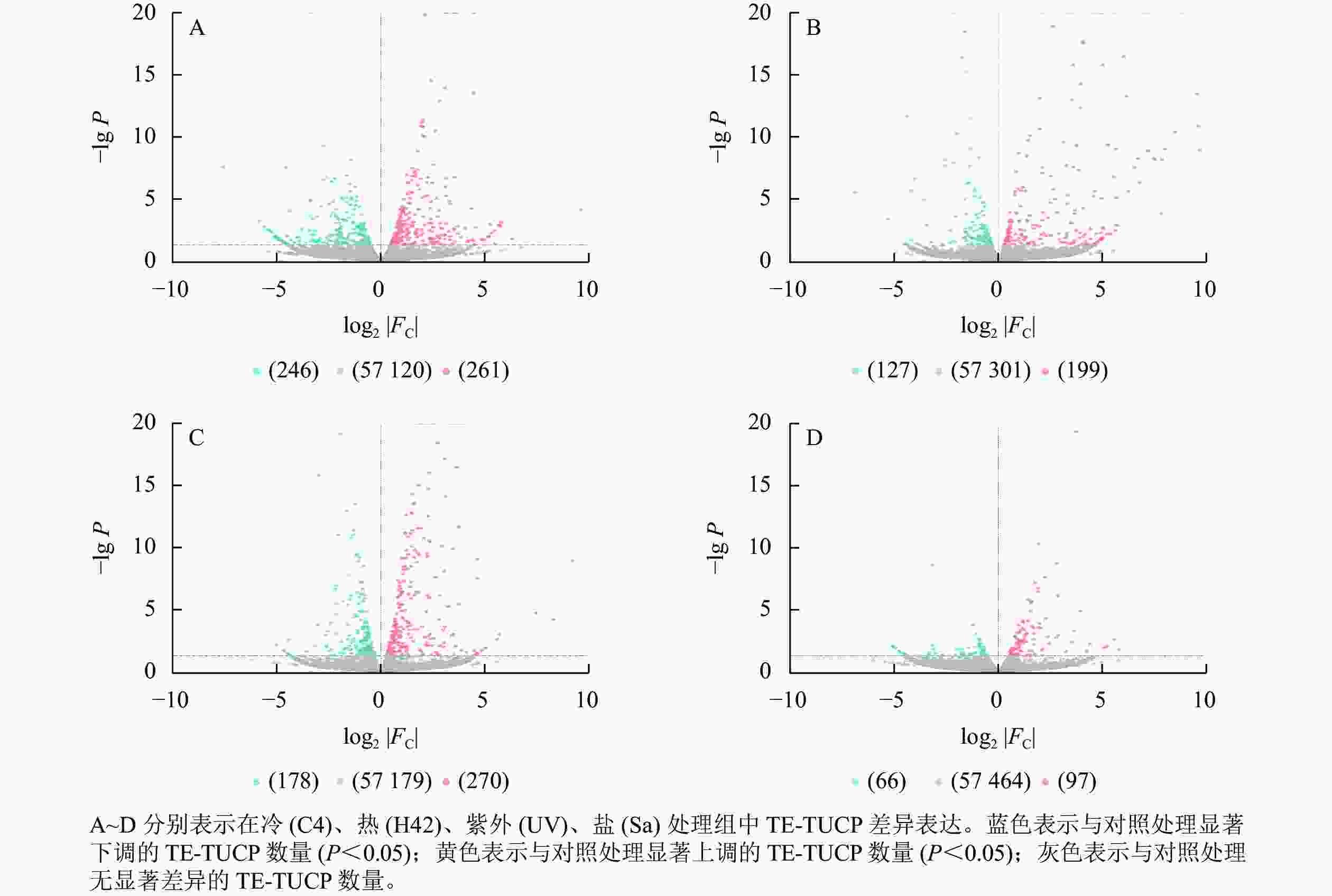
图 2 毛竹TE-TUCP表达模式分析
Figure 2. Analysis of TE-TUCP expression pattern in Ph. edulis
-
对TUCP-TE附近4 646个基因研究发现:C4与ck处理组共筛选出758个上调基因,833个下调基因(图3A),差异表达基因占所有附近基因的34.24%。H42与ck处理组共筛选出358个上调基因,334个下调基因(图3B),差异表达基因占所有附近基因的14.89%。UV与ck处理组共筛选出739个上调基因,596个下调基因(图3C),差异表达基因占所有附近基因的28.73%。Sa与wa处理组共筛选出320个上调基因,201个下调基因(图3D),差异表达基因占所有附近基因的11.21%。
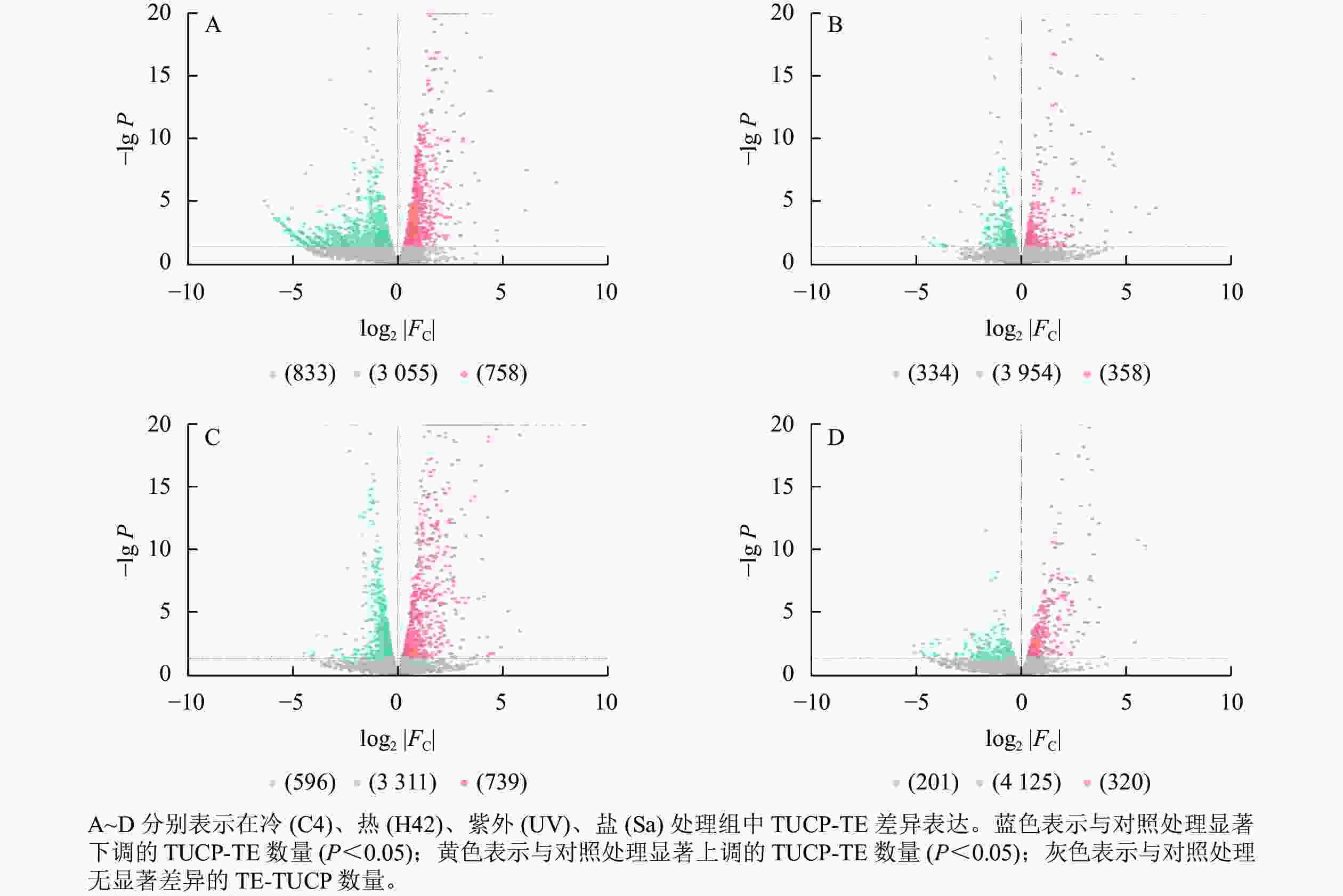
图 3 毛竹TUCP-TE附近基因表达模式分析
Figure 3. Analysis of gene expression patterns in the vicinity of Ph. edulis TUCP-TE
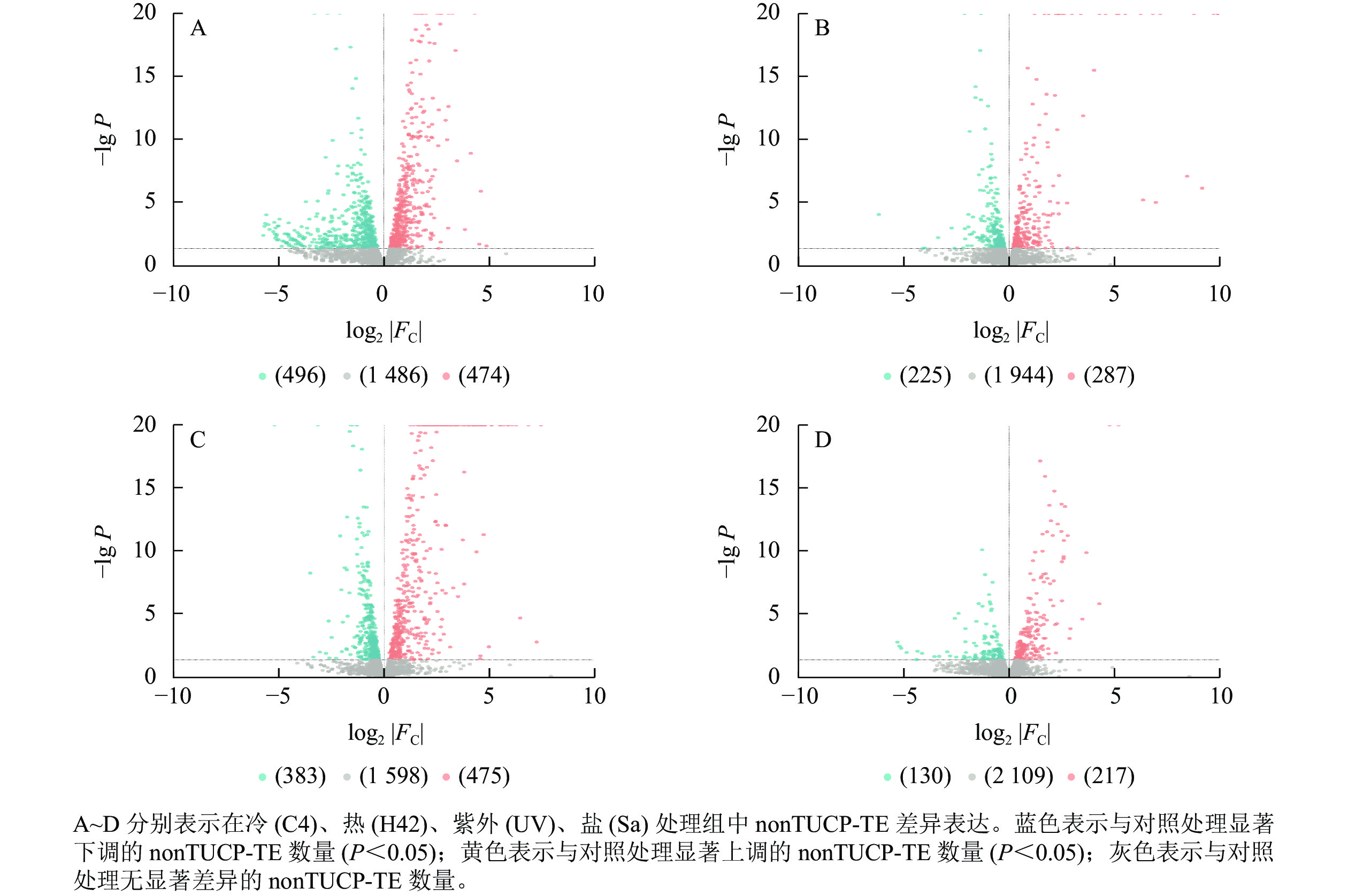
对nonTUCP-TE附近2 456个基因研究发现:C4与ck处理组共筛选出474个上调基因,496个下调基因(图4A),差异表达基因占所有附近基因的39.50%。H42与ck处理组共筛选出287个上调基因,225个下调基因(图4B),差异表达基因占所有附近基因的20.85%。UV与ck处理组共筛选出475个上调基因,383个下调基因(图4C),差异表达基因占所有附近基因的34.93%。Sa与wa处理组共筛选出217个上调基因,130个下调基因(图4D),差异表达基因占所有附近基因的14.13%。可见,除了低温处理外,高温、高盐、紫外照射处理组TUCP-TE附近5 000 bp距离内基因差异表达数量均比nonTUCP-TE高,表明高温、高盐、紫外照射可以促进TUCP-TE附近差异基因表达,但是低温会抑制TUCP-TE附近差异基因表达。
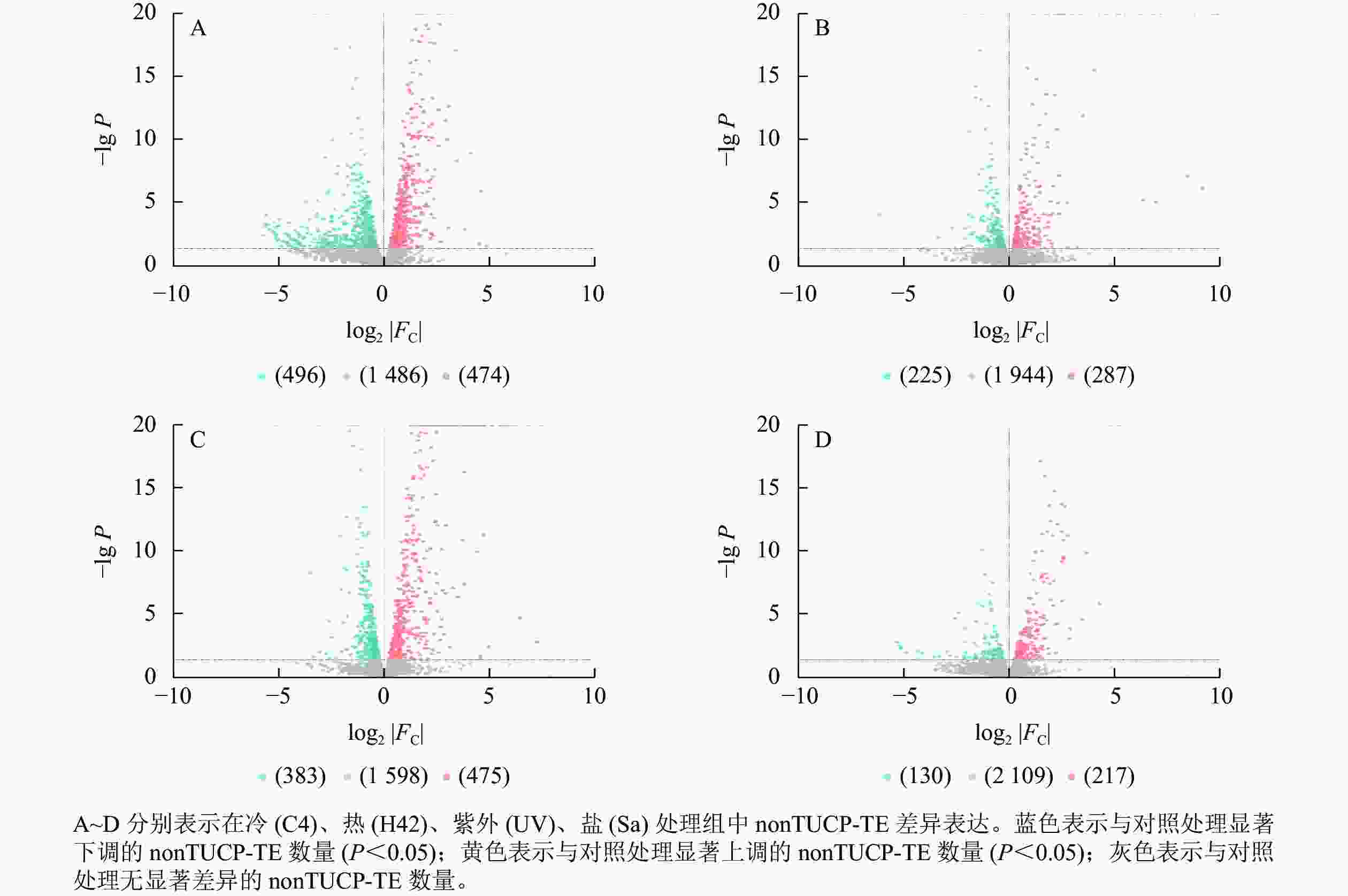
图 4 毛竹nonTUCP-TE附近基因表达模式分析
Figure 4. Analysis of gene expression patterns in the vicinity of Ph. edulis nonTUCP-TE
-
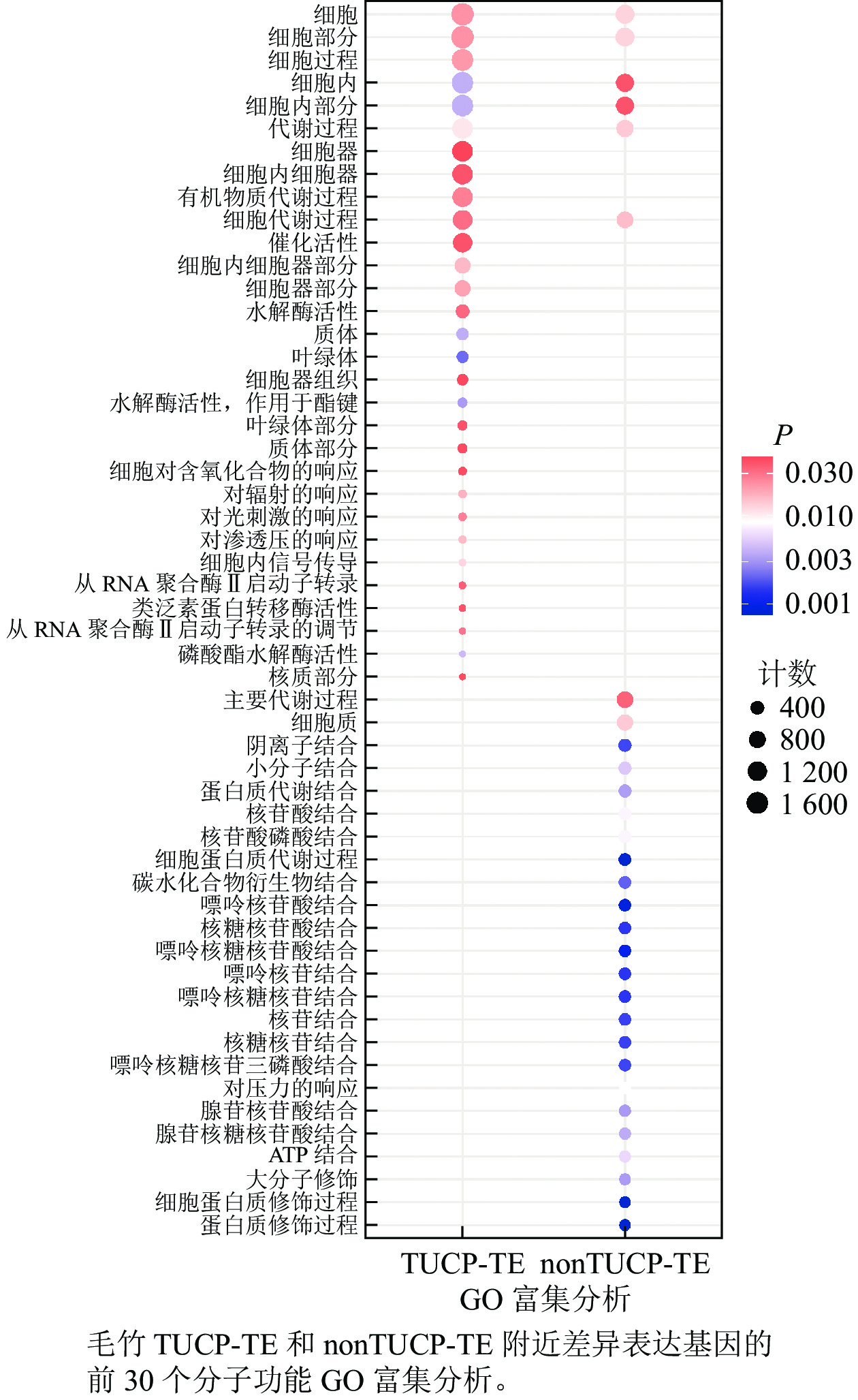
在附近5 000 bp距离内,分别取TUCP-TE和nonTUCP-TE差异表达基因合集2 592、1 591个基因进行GO富集分析。如图5所示:与nonTUCP-TE相比,非生物胁迫下,TUCP-TE附近5 000 bp距离内差异表达基因,除了共同显著富集在细胞、细胞组分、细胞内、细胞内组分、代谢过程外,还额外显著富集在与非生物胁迫相关的催化活性、水解酶活性、对辐射反应、对光刺激反应、对渗透胁迫的反应等方面。表明非生物胁迫可以促进毛竹TUCP-TE附近相关基因的表达。
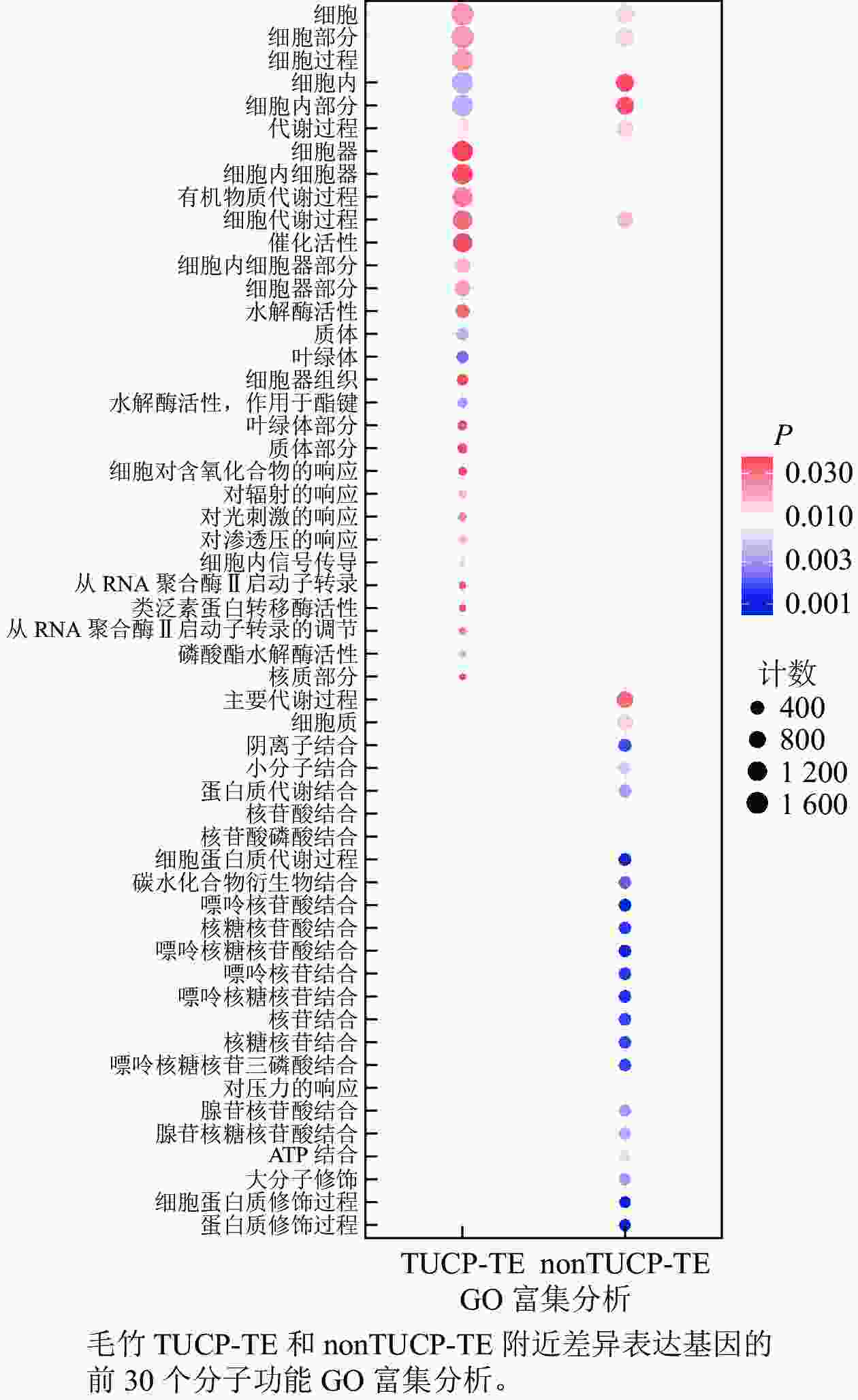
图 5 GO富集分析
Figure 5. GO enrichment analysis
-
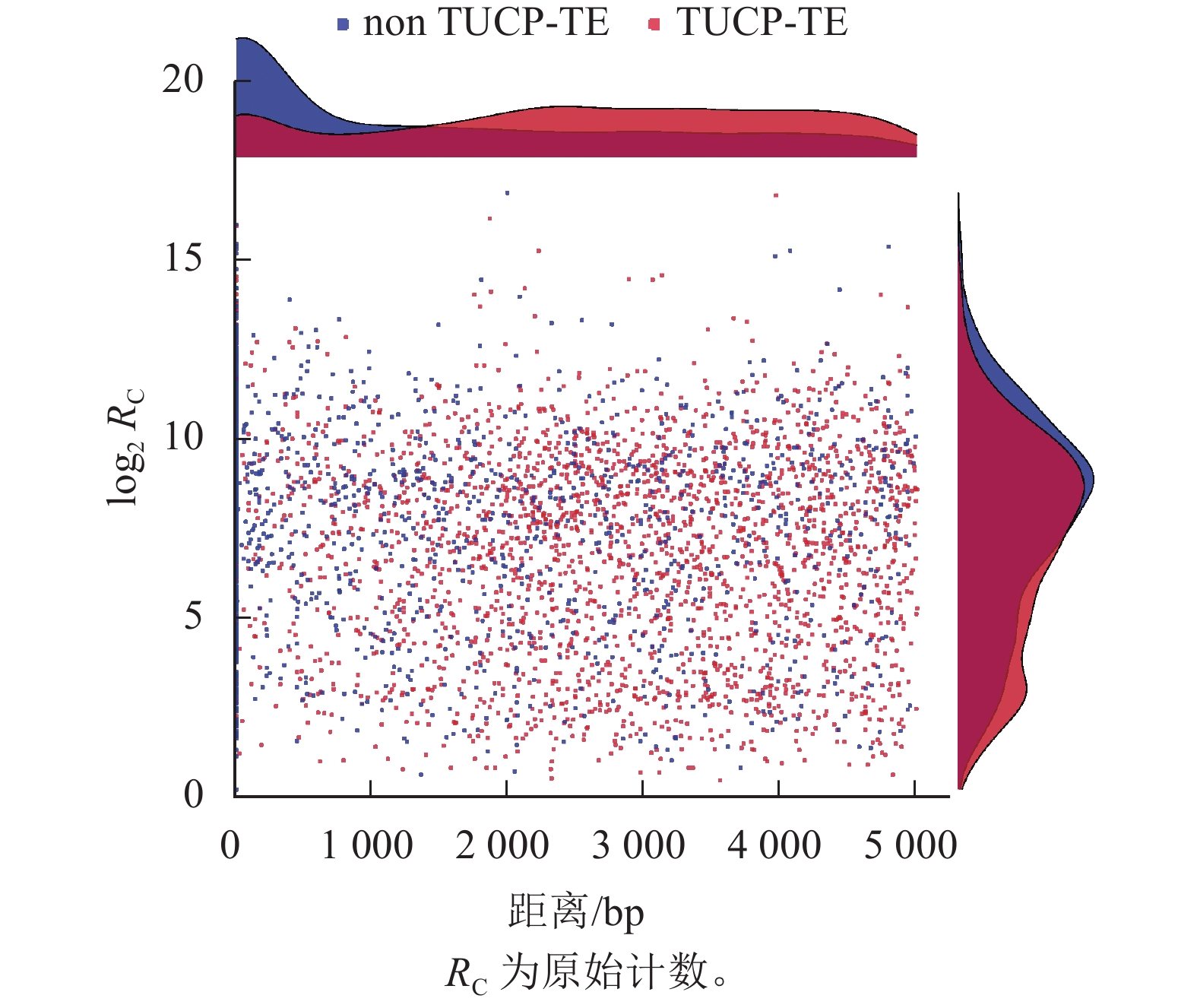
如图6所示:nonTUCP-TE附近1 500 bp内的差异表达基因比TUCP-TE相同距离内差异表达基因的数量多,且在越靠近nonTUCP-TE的位置,差异表达基因集中。在附近1 500 bp内,TUCP-TE附近越靠近TUCP,差异表达基因就数量越少,而在距离2 000~3 000 bp内,TUCP-TE附近差异表达基因数量较多。说明基因的表达潜能与邻近TE-TUCP的表达潜能互相抑制。2 000~3 000 bp是TE-TUCP对基因影响较明显的范围。同时,TUCP-TE附近差异表达基因平均表达量为757.1,低于nonTUCP-TE (1 245.5)。TUCP-TE附近差异表达基因表达水平与nonTUCP-TE相比,整体水平上表达峰更低,说明非生物胁迫可能会抑制TE-TUCP附近基因的整体表达水平。

图 6 TE-TUCP与基因距离、基因表达水平统计散点图
Figure 6. Scatterplot of TE-TUCP versus gene distance and gene expression level statistics
-
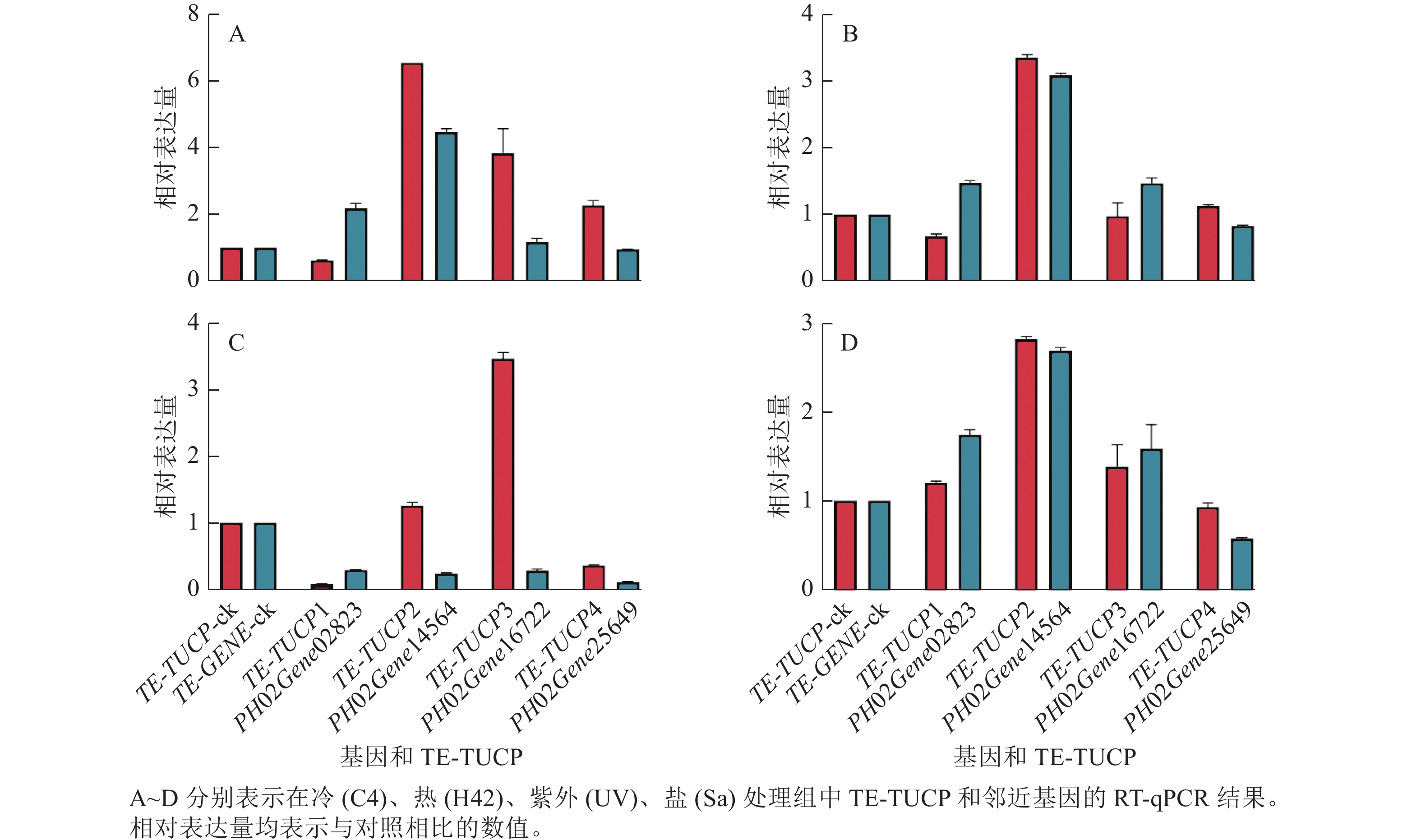
为了验证转录组测序数据分析结果的准确性,从鉴定的差异表达TE-TUCP中随机挑选4个TE-TUCP及其附近基因,分别是TE-TUCP1和PH02Gene02823、TE-TUCP2和PH02Gene14564、TE-TUCP3和PH02Gene16722,以及TE-TUCP4和PH02Gene25649。引物见表2。如图7所示:TE-TUCP和邻近基因的RT-qPCR结果与转录组分析结果一致,说明转录组测序数据分析结果是可靠的。
表 2 引物序列信息
Table 2. Primer sequence information
引物名称 序列(5′→3′) 引物名称 序列(5′→3′) TE-TUCP1-F AACAAGGCAGCGCAGCAGAC TE-TUCP3-R AACTAATGGAAGCGGACGCACG TE-TUCP1-R TTGGCGGCACCTTAGGCTGA PH02Gene16722-F CGAATGGCAGGAGGAGCAGAGA PH02Gene02823-F CTCCACGCCCATCAACACCAAG PH02Gene16722-R TCTTGCCCTTGCCGAAGTGGA PH02Gene02823-R ACTGAGGAGGGAGGAGGCAACT TE-TUCP4-F AGGCAGATTCCGCAGGTGGTT TE-TUCP2-F ATGGTGTTGGTGGTGTGCGTG TE-TUCP4-R ATTCACCAGCATCCAGCTTGGC TE-TUCP2-R CGGCAGATTGCGTGCGTACATA PH02Gene25649-F AATTGCACCTGCCTGCTGGATG PH02Gene14564-F GGAAGGTCAGGCACCAACGATG PH02Gene25649-R ACCTCCCGTCACTGGTCCTTTG PH02Gene14564-R AGCCACCACTGCTACCGTAGTC Actin-F ATACGCTTCCTCACGCTATTCTT TE-TUCP3-F AGCCACGGATTCAGCAACAAGG Actin-R CCGAGCTTCTCCTTTATGTCCCT 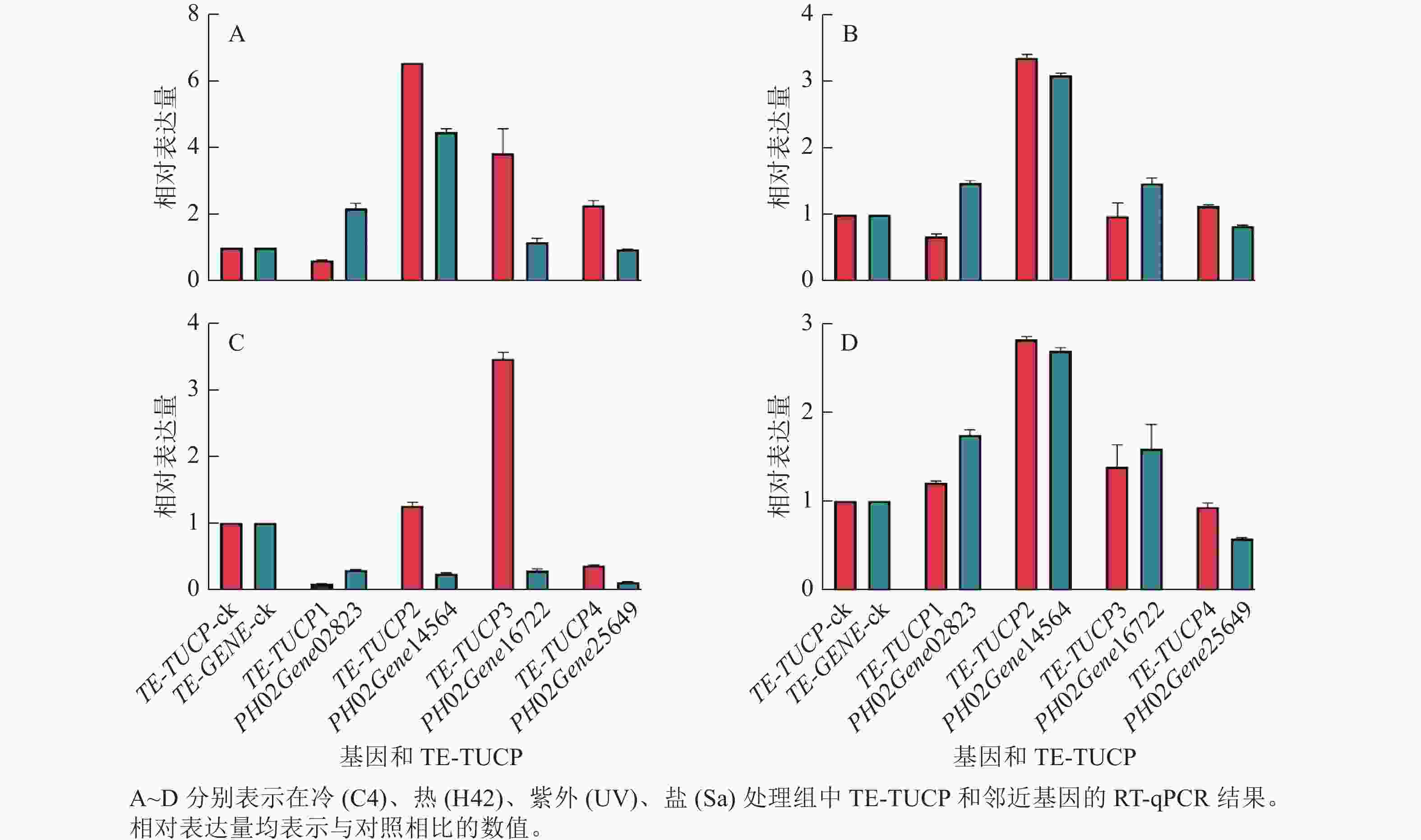
图 7 4个TE-TUCP及其邻近4个基因的RT-qPCR表达分析
Figure 7. RT-qPCR expression analysis of 4 TE-TUCP and four nearby genes
-
植物基因组中转座子含量丰富,毛竹基因组共鉴定出TE序列991 865条,占基因组大小的63%[3]。在桑树Morus alba基因组共检测到TE序列286 122条,占基因组大小的32%[38],玉米基因组中转座子占比70%[39],高粱Sorghum bicolor基因组中转座子占比55%[40],水稻基因组中转座子占比26%[41]。可见,TE在植物基因组中占比较高。
本研究表明:毛竹共鉴定出87 409个TUCP,它们的平均长度比mRNA的平均长度短,但比lncRNA的平均长度长。TUCP、mRNA和lncRNA之间的ORF长度具有相同的规律。这与其他植物研究结果相似[14, 17]。
含有应激响应顺式调控元件的TE通过表观遗传修饰对环境胁迫表现出快速反应。例如,番茄Solanum lycopersicum基因组中属于Ty1/Copia超家族的反转录转座子在干旱胁迫和脱落酸(ABA)作用下被激活[42]。番茄和烟草Nicotiana tabacum中的TE可被低温特异性触发和激活[43]。以上研究表明:TE衍生的TUCP能在非生物胁迫下调控自身转录和附近基因的表达。毛竹57 627个TE-TUCP,65.9%来自TE。TE-TUCP主要来源于Ty1/Copia和Ty3/Gypsy超家族。TE-TUCP表现出胁迫特异性表达模式,在冷、盐胁迫下下调数量大于上调数量,在热胁迫和紫外胁迫下上调数量大于下调数量。
本研究发现:低温胁迫抑制TUCP-TE附近基因差异表达,高盐胁迫促进TUCP-TE附近基因差异表达,TE-TUCP与附近基因的表达潜能互相抑制,这与之前拟南芥中TUCP与附近基因转录水平呈负相关结果一致[19]。另外,高温胁迫也能促进TUCP-TE附近基因差异表达。红枣转录组研究表明:TUCP改变了附近基因表达以适应高温环境[20]。本研究还发现:紫外胁迫下,TE-TUCP促进光刺激反应相关基因差异表达。棉花中TUCP参与光合作用并影响生长发育过程[14]。GO富集分析结果显示:TE-TUCP通过调控关键基因在非生物胁迫反应中发挥重要作用。这与TE在非生物胁迫下可重新激活转录从而应对环境条件变化的结论相一致[44]。
-
本研究在4种胁迫处理的转录组数据中,共识别出57 627个毛竹TE-TUCP。这些TE-TUCP在面对不同的非生物胁迫时,展现出了各自独特的表达模式。它们主要源自Ty1/Copia和Ty3/Gypsy这2个超家族。基因自身的表达潜力与邻近的TE-TUCP的表达潜力之间存在着一种相互抑制的现象。此外,非生物胁迫还会通过调控TE-TUCP的转录情况,来影响周边基因的表达,从而帮助毛竹适应这些胁迫条件。
Effects of abiotic stress treatments on the transcriptional activity of transposable element-derived TUCP in Phyllostachys edulis
-
摘要:
目的 转座子(TE)是真核细胞基因组的重要组成部分,在毛竹Phyllostachys edulis基因组超过63%时,易受胁迫诱导激活。分析非生物胁迫下,来源于转座子的不确定编码潜力转录本(TUCP)的表达模式,为转座子参与毛竹抗逆分子机制提供参考。 方法 采用生物信息学技术和手段,在低温、高温、高盐、紫外照射等4种胁迫处理下,研究毛竹TE-TUCPs及转座子邻近基因的转录特性和转录模式。通过实时荧光定量PCR (RT-qPCR)验证转录组来源的TE-TUCPs差异表达数据的可靠性。 结果 在毛竹4个胁迫处理转录本中,共鉴定出57 627个TE-TUCPs。TE-TUCPs应对不同非生物胁迫表现出特异性表达模式。高温、高盐、紫外照射处理可以促进具有转录活性的TE-TUCPs附近基因差异表达,但是低温会抑制具有转录活性的TE-TUCPs附近基因差异表达。 结论 TE-TUCPs主要来源于Ty1/Copia和Ty3/Gypsy超家族。基因的表达潜能与近距离的TE-TUCPs表达潜能互相抑制。TE-TUCPs转录情况会受到非生物胁迫作用来调控附近基因的表达以适应胁迫影响。图7表2参44 -
关键词:
- 毛竹 /
- 转座子(TE) /
- 不确定编码潜力转录本(TUCP) /
- 非生物胁迫
Abstract:Objective Transposable elements (TE), an essential component of eukaryotic genomes are prone to activation under stress when they account for over 63% of the Phyllostachys edulis genome. This study, with an analysis of the expression patterns of transcripts of uncertain coding potential (TUCP) from transposable elements under abiotic stress, is aimed to provide insights into the molecular mechanisms of TEs in stress resistance in Ph. edulis. Method First, bioinformatics techniques were employed to investigate the transcriptional characteristics and patterns of TE-TUCPs, and neighboring genes in Ph. edulis under 4 stress treatments: low temperature, high temperature, high salinity, and UV irradiation. Then the reliability of the differentially expressed TE-TUCPs, data derived from the transcriptome was validated using RT-qPCR. Result A total of 57 627 TE-TUCPs were identified from the transcripts of Ph. edulis under 4 stress treatments. These TE-TUCPs exhibited specific expression patterns in response to different abiotic stresses. High temperature, high salinity, and UV irradiation promoted differential expression of genes neighboring TE-TUCPs with transcriptional activity, whereas low temperature suppressed such differential expression. Conclusion TE-TUCPs were primarily derived from the Ty1/Copia and Ty3/Gypsy superfamilies. The expression potential of genes and that of nearby TE-TUCPs were mutually inhibitory and the transcription of TE-TUCPs was regulated by abiotic stress to modulate the expression of neighboring genes in response to stress. [Ch, 7 fig. 2 tab. 44 ref.] -
图 3 毛竹TUCP-TE附近基因表达模式分析
Figure 3 Analysis of gene expression patterns in the vicinity of Ph. edulis TUCP-TE
图 4 毛竹nonTUCP-TE附近基因表达模式分析
Figure 4 Analysis of gene expression patterns in the vicinity of Ph. edulis nonTUCP-TE
图 6 TE-TUCP与基因距离、基因表达水平统计散点图
Figure 6 Scatterplot of TE-TUCP versus gene distance and gene expression level statistics
图 7 4个TE-TUCP及其邻近4个基因的RT-qPCR表达分析
Figure 7 RT-qPCR expression analysis of 4 TE-TUCP and four nearby genes
表 1 毛竹TE-TUCP转座子来源
Table 1. Sources of TE-TUCP transposons in Ph. edulis
转座子来源分类 TUCP-TE/
个nonTUCP-TE/
个数量合
计/个DNA 45 115 160 DNA/CMC-EnSpm 2 994 1 516 4 510 DNA/hAT 4 18 22 DNA/hAT-Ac 745 907 1 652 DNA/hAT-Tag1 17 26 43 DNA/hAT-Tip100 228 209 437 DNA/MULE-MuDR 2 368 1 894 4 262 DNA/PIF-Harbinger 112 154 266 DNA/TcMar-Stowaway 153 452 605 LINE/L1 1 040 1 050 2 090 Low_complexity 4 9 13 LTR 68 1058 1126 LTR/Caulimovirus 6 39 45 LTR/Copia 8 456 8 809 17 265 LTR/Gypsy 10 662 13 318 23 980 Other/centromeric 2 2 4 RC/Helitron 241 427 668 Simple_repeat 112 341 453 SINE/L1 6 6 12 SINE 2 2 未知 12 12 合计 27 263 30 364 57 627  下载: 导出CSV
下载: 导出CSV
表 2 引物序列信息
Table 2. Primer sequence information
引物名称 序列(5′→3′) 引物名称 序列(5′→3′) TE-TUCP1-F AACAAGGCAGCGCAGCAGAC TE-TUCP3-R AACTAATGGAAGCGGACGCACG TE-TUCP1-R TTGGCGGCACCTTAGGCTGA PH02Gene16722-F CGAATGGCAGGAGGAGCAGAGA PH02Gene02823-F CTCCACGCCCATCAACACCAAG PH02Gene16722-R TCTTGCCCTTGCCGAAGTGGA PH02Gene02823-R ACTGAGGAGGGAGGAGGCAACT TE-TUCP4-F AGGCAGATTCCGCAGGTGGTT TE-TUCP2-F ATGGTGTTGGTGGTGTGCGTG TE-TUCP4-R ATTCACCAGCATCCAGCTTGGC TE-TUCP2-R CGGCAGATTGCGTGCGTACATA PH02Gene25649-F AATTGCACCTGCCTGCTGGATG PH02Gene14564-F GGAAGGTCAGGCACCAACGATG PH02Gene25649-R ACCTCCCGTCACTGGTCCTTTG PH02Gene14564-R AGCCACCACTGCTACCGTAGTC Actin-F ATACGCTTCCTCACGCTATTCTT TE-TUCP3-F AGCCACGGATTCAGCAACAAGG Actin-R CCGAGCTTCTCCTTTATGTCCCT
下载: 导出CSV
-
[1] LIU Yuli, ZHOU Guomo, DU Huaqiang, et al. Soil respiration of a moso bamboo forest significantly affected by gross ecosystem productivity and leaf area index in an extreme drought event [J/OL]. PeerJ, 2018, 6: e5747[2024-02-20]. doi: 10.7717/peerj.5747. [2] RAMAKRISHNAN M, YRJÄLÄ K, VINOD K K, et al. Genetics and genomics of moso bamboo (Phyllostachys edulis): current status, future challenges, and biotechnological opportunities toward a sustainable bamboo industry [J/OL]. Food and Energy Security, 2020, 9(4): e229[2024-02-20]. doi: 10.1002/fes3.229. [3] ZHAO Hansheng, GAO Zhimin, WANG Le, et al. Chromosome-level reference genome and alternative splicing atlas of moso bamboo (Phyllostachys edulis) [J/OL]. GigaScience, 2018, 7(10): giy115[2024-02-20]. doi: 10.1093/gigascience/giy115. [4] BOURQUE G, BURNS K H, GEHRING M, et al. Ten things you should know about transposable elements [J/OL]. Genome Biology, 2018, 19: 199[2024-02-20]. doi: 10.1186/s13059-018-1577-z. [5] RAMAKRISHNAN M, PAPOLU P K, MULLASSERI S, et al. The role of LTR retro transposons in plant genetic engineering: how to control their transposition in the genome [J]. Plant Cell Reports, 2023, 42(1): 3 − 15. [6] LIU Beibei, Zhao Meixia. How transposable elements are recognized and epigenetically silenced in plants? [J/OL]. Current Opinion in Plant Biology, 2023, 75: 102428[2024-02-20]. doi: 10.1016/j.pbi.2023.102428. [7] RAMAKRISHNAN M, SATISH L, KALENDAR R, et al. The dynamism of transposon methylation for plant development and stress adaptation [J/OL]. International Journal of Molecular Sciences, 2021, 22(21): 11387[2024-02-20]. doi: 10.3390/ijms222111387. [8] NAITO K, FENG Zhang, TSUKIYAMA T, et al. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression [J]. Nature, 2009, 461(7267): 1130 − 1134. [9] ZHOU Mingbing, ZHU Yihang, BAI Youhuang, et al. Transcriptionally active LTR retroelement-related sequences and their relationship with small RNA in moso bamboo (Phyllostachys edulis) [J/OL]. Molecular Breeding, 2017, 37(10): 132[2024-02-20]. doi: 10.1007/s11032-017-0733-6. [10] SUONIEMI A, NARVANTO A, SCHULMAN A H. The BARE-1 retrotransposon is transcribed in barley from an LTR promoter active in transient assays [J]. Plant Molecular Biology, 1996, 31(2): 295 − 306. [11] CABILI M N, TRAPNELL C, GOFF L, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses [J]. Genes &Development, 2011, 25(18): 1915 − 1942. [12] WASHIETL S, KELLIS M, GARBER M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals [J]. Genome Research, 2014, 24(4): 616 − 628. [13] KONDO T, PLAZA S, ZANET J, et al. Small peptides switch the transcriptional activity of shavenbaby during drosophila embryogenesis [J]. Science, 2010, 329(5989): 336 − 339. [14] SALIH H, GONG Wengfang, HE Shoupu, et al. Comparative transcriptome analysis of TUCPs in Gossypium hirsutum ligon-lintless-1 mutant and their proposed functions in cotton fiber development [J]. Molecular Genetics and Genomics, 2019, 294(1): 23 − 34. [15] XIAO Ke, YANG Yuemei, BIAN Yanyan, et al. Identification of differentially expressed long noncoding RNAs in human knee osteoarthritis [J]. Journal of Cellular Biochemistry, 2019, 120(3): 4620 − 4633. [16] LUO Honglin, YANG Huizan, LIN Yong, et al. LncRNA and mRNA profiling during activation of tilapia macrophages by HSP70 and antigen [J]. Oncotarget, 2017, 8(58): 98455 − 98470. [17] LIU Yong, QI Bing, XIE Juan, et al. Filtered reproductive long non-coding RNAs by genome-wide analyses of goat ovary at different estrus periods [J]. BMC Genomics, 2018, 19(1): 866[2024-02-20]. doi:10.1186/s12864-018-5268-7. [18] LIU Huimin, LU Yan, WANG Juan, et al. Genome-wide screening of long non-coding RNAs involved in rubber biosynthesis [J]. Journal of Integrative Plant Biology, 2018, 60(11): 1070 − 1082. [19] WANG Dong, QU Zhipeng, YANG Lan, et al. Transposable elements (TEs) contribute to stress-related long intergenic noncoding RNAs in plants [J]. Plant Journal, 2017, 90(1): 133 − 146. [20] HAO Qin, YANG Lei, FAN Dingyu, et al. The transcriptomic response to heat stress of a jujube (Ziziphus jujuba Mill. ) cultivar is featured with changed expression of long noncoding RNAs [J/OL]. PLoS One, 2021, 16(5): e0249663[2024-02-20]. doi: 10.1371/journal.pone.0249663. [21] DING Yiqian, ZOU Longhai, WU Jiajun, et al. The pattern of DNA methylation alteration, and its association with the expression changes of non-coding RNAs and mRNAs in moso bamboo under abiotic stress [J/OL]. Plant Science, 2022, 325: 111451[2024-02-20]. doi: 10.1016/j.plantsci.2022.111451. [22] BOLGER A M, LOHSE M, USADEL B. Trimmomatic: a flexible trimmer for Illumina sequence data [J]. Bioinformatics, 2014, 30(15): 2114 − 2134. [23] KIM D, PAGGI J M, PARK C, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype [J]. Nature Biotechnology, 2019, 37(8): 907 − 915. [24] LIAO Yang, SMYTH G K, SHI Wei. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features [J]. Bioinformatics, 2014, 30(7): 923 − 930. [25] LOVE M I, HUBER W, ANDERS S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 [J/OL]. Genome Biology, 2014, 15: 550[2024-02-20]. doi: 10.1186/s13059-014-0550-8. [26] TRAPNELL C, ROBERTS A, GOFF L, et al. Differential gene and transcript expression analysis of RNA-Seq experiments with TopHat and Cufflinks [J]. Nature Protocols, 2012, 7(3): 562 − 578. [27] SUN Liang, LUO Haitao, BU Dechao, et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts [J/OL]. Nucleic Acids Research, 2013, 41(17): e166[2024-02-20]. doi: 10.1093/nar/gkt646. [28] MISTRY J, BATEMAN A, FINN R D. Predicting active site residue annotations in the Pfam database [J/OL]. BMC Bioinformatics, 2007, 8: 298[2024-02-20]. doi:10.1186/1471-2105-8-298. [29] KANG Yujian, YANG Dechang, KONG Lei, et al. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features [J]. Nucleic Acids Research, 2017, 45(W1): W12 − W16. [30] ZHOU Mingbing, TAO Guiyun, PI Peiyao, et al. Genome-wide characterization and evolution analysis of miniature inverted-repeat transposable elements (MITEs) in moso bamboo (Phyllostachys heterocycla) [J]. Planta, 2016, 244(4): 775 − 787. [31] CHEN Nansheng. Using RepeatMasker to identify repetitive elements in genomic sequences [J]. Current Protocols in Bioinformatics, 2004, 5(1): 4 − 10. [32] QUINLAN A R, HALL I M. BEDTools: a flexible suite of utilities for comparing genomic features [J]. Bioinformatics, 2010, 26(6): 841 − 842. [33] PERTEA M, PERTEA G M, ANTONESCU C M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads [J]. Nature Biotechnology, 2015, 33(3): 290 − 295. [34] PERTEA G, PERTEA M. GFF Utilities: GffRead and GffCompare [J/OL]. F1000Research, 2020, 9: 304[2024-02-20]. doi: 10.12688/f1000research.23297.2. [35] SONESON C, LOVE M I, ROBINSON M D. Differential analyses for RNA-Seq: transcript-level estimates improve gene-level inferences [J/OL]. F1000Research, 2015, 4: 1521[2024-02-20]. doi: 10.12688/f1000research.7563.2. [36] LIVAK K J, SCHMITTGEN T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method [J]. Methods, 2001, 25(4): 402 − 408. [37] 陈娅欣, 周明兵. 毛竹长末端重复序列反转录转座子的全基因组特征及进化分析[J]. 浙江农林大学学报, 2021, 38(3): 455 − 463. CHEN Yaxin, ZHOU Mingbing. Genome-wide characteristics and evolution analysis of long terminal repeat retrotransposons in Phyllostachys edulis [J]. Journal of Zhejiang A&F University, 2021, 38(3): 455 − 463. [38] XIN Youchao, MA Bi, XIANG Zhonghua, et al. Amplification of miniature inverted-repeat transposable elements and the associated impact on gene regulation and alternative splicing in mulberry (Morus notabilis) [J/OL]. Mobile DNA, 2019, 10: 27[2024-02-20]. doi: 10.1186/s13100-019-0169-0. [39] XU Ling, ZHANG Yu, SU Yuan, et al. Structure and evolution of full-length LTR retrotransposons in rice genome [J]. Plant Systematics and Evolution, 2010, 287(1/2): 19 − 28. [40] PATERSON A H, BOWERS J E, BRUGGMANN R, et al. The Sorghum bicolor genome and the diversification of grasses [J]. Nature, 2009, 457(7229): 551 − 556. [41] WANG Hao, XU Zhao, YU Hongjie. LTR retrotransposons reveal recent extensive inter-subspecies nonreciprocal recombination in Asian cultivated rice [J/OL]. BMC Genomics, 2008, 9(1): 565[2024-02-20]. doi: 10.1186/1471-2164-9-565. [42] SALLAM N, MOUSSA M. DNA methylation changes stimulated by drought stress in ABA-deficient maize mutant [J]. Plant Physiology and Biochemistry, 2021, 160: 218 − 224. [43] BENOIT M, DROST H G, CATONI M, et al. Environmental and epigenetic regulation of retrotransposons in tomato [J/OL]. PLoS Genetics, 2019, 15(9): e1008370[2024-02-20]. doi: 10.1371/journal.pgen.1008370. [44] CASACUBERTA E, GONZÁLEZ J. The impact of transposable elements in environmental adaptation [J]. Molecular Ecology, 2013, 22(6): 1503 − 1517. -
-
链接本文:
https://zlxb.zafu.edu.cn/article/doi/10.11833/j.issn.2095-0756.20240195
 点击查看大图
点击查看大图
计量
- 文章访问数: 1499
- HTML全文浏览量: 492
- PDF下载量: 36
- 被引次数: 0
