





-
小反刍兽疫(peste des petits ruminants,PPR)是由小反刍兽疫病毒(peste des petits ruminants virus,PPRV)引起的一种急性病毒性传染病,主要感染山羊Capra hircus、绵羊Ovis aries以及其他野生小反刍兽,以发热,眼、鼻分泌物增多,胃炎、腹泻和肺炎为特征。该病主要流行于亚洲和非洲,2007年中国在西藏阿里地区首次发现该病,2010年8月在阿里地区日土县再次发生疫情[1-2]。2013年末,该病传入中国新疆地区,并迅速传播至22个省份,对中国养羊业造成严重危害[3-4]。PPRV属于副黏病毒科Paramyxovirdae麻疹病毒属Morbolivirus;基因组为单股负链RNA,长度为15 948 nt或者15 954 nt;基因组3′末端为前导序列(leader),5′末端为尾随序列(trailer);6个基因排列顺序为3′-N-P-M-F-H-L-5′,依次编码6个结构蛋白:核衣壳蛋白(N)、磷蛋白(P)、基质蛋白(M)、融合蛋白(F)、血凝蛋白(H)和大蛋白(L);P基因还编码2个非结构蛋白C和V。根据F基因或N基因部分核苷酸序列差异可将PPRV毒株分为4个基因系[5-6],亚洲国家流行的毒株属于基因Ⅳ系[1]。小反刍兽疫病毒在流行过程中,基因组发生点突变,核苷酸变异速率大约为9×10−4位点·a−1[7-8]。对中国2013年11月至2014年6月间25个毒株的基因组序列的比对和分析发现:P基因变异最大,非同义突变率最高[8]。随着全面免疫的实施,受免疫选择压的作用,病毒各基因尤其是P基因的变异率及非同义突变率发生变化,有必要进一步研究。PPRV P基因核苷酸长度为1 530 nt,编码509个氨基酸,编码的P蛋白分子量约54.9 kD。P蛋白在病毒RNA的转录和复制过程中发挥作用,也能单独与N蛋白-RNA模板复合物结合激活转录,同时与L蛋白相结合形成依赖于RNA的RNA聚合酶,并与N蛋白-RNA模板结合形成核糖核蛋白复合体,进行病毒RNA的转录和复制[9]。本研究对2013年以来中国PPRV流行毒株P基因进行序列测定和分析,探讨病毒流行过程中P基因的序列变异情况。
-
2014−2017年,采集到9个省12个疫点的PPRV阳性组织样品12份,由中国动物卫生与流行病学中心保存并提供,具体样品信息见表1。
表 1 2014−2017年中国12株PPRV毒株信息
Table 1. Details of the 12 PPRV strains collected in China during 2014−2017
毒株编号 采样时间(年-月-日) 来源省份 毒株编号 采样时间(年-月-日) 来源省份 JL472014 2014-04-06 吉林 JS142016 2016-01-20 江苏 JX62014 2014-07-21 江西 GX22016 2016-08-22 广西 ZJ82014 2014-10-11 浙江 HN182016 2016-10-16 湖南 JX172015 2015-03-30 江西 XJ302017 2017-02-12 新疆 AH162015 2015-04-09 安徽 HN02017 2017-03-05 湖南 GZ112015 2015-09-02 贵州 GZ252017 2017-09-21 贵州 -
取肠系淋巴结组织100 mg,用组织匀浆仪匀浆后,根据High Pure Viral RNA Kit (Roche)的操作说明提取病毒RNA,−80 ℃保存备用。
-
采用Prime ScriptTM One Step RT-PCR Kit (TaKaRa)扩增PPRV P基因。反应体系共50 µL,包括2× 1 Step Buffer 25 µL,Primerscript 1 step Enzyme Mix 2 µL,上、下游引物各2 µL,病毒RNA 5 µL。RT-PCR反应条件为:50 ℃逆转录30 min,94 ℃ 2 min,进行Taq酶激活;40个循环的聚合酶链式反应(PCR)(94 ℃ 1 min,68 ℃ 1 min,72 ℃ 4 min);72 ℃ 7 min再延伸。对PCR产物进行切胶回收。
-
纯化后的PCR产物送青岛华大公司测序,获得12个毒株的P基因序列;从GenBank中下载2013−2014年中国25个PPRV流行毒株的基因组序列[8](表2),截取P基因序列后进行核苷酸和氨基酸序列比对。从GenBank中下载中国及其他10个国家共15个PPRV代表毒株的基因组序列(表3),绘制系统进化树。用MEGA 4.0 软件进行序列比对,用最大似然法构建分子进化树,bootstrap测试1 000次重复。用Simplot软件进行序列同源性分析。
表 2 2013−2014年中国PPRV流行毒株信息
Table 2. Details of the PPRV strains collected in China during 2013−2014
病毒名称 采样时间(年-月-日) 来源省份 宿主 病毒名称 采样时间(年-月-日) 来源省份 宿主 XJYL2013 2013-11-30 新疆 山羊 JL2014 2014-04-01 吉林 绵羊 XJ22013 2013-12-20 新疆 山羊 JS2014 2014-04-02 江苏 山羊 XJ32013 2013-12-21 新疆 绵羊 HeN2014 2014-04-03 河南 山羊 XJ42013 2013-12-22 新疆 山羊 HB2014 2014-04-03 湖北 山羊 XJ52013 2013-12-29 新疆 山羊 AH2014 2014-04-03 安徽 山羊 GS2014 2014-01-22 甘肃 绵羊 SX2014 2014-04-05 山西 山羊 NX2014 2014-02-17 宁夏 绵羊 GX2014 2014-04-16 广西 山羊 LN2014 2014-03-17 辽宁 山羊 GZ2014 2014-04-21 贵州 山羊 CQ2014 2014-03-30 重庆 山羊 ZJ2014 2014-04-25 浙江 山羊 HLJ2014 2014-03-31 黑龙江 山羊 HN2014 2014-04-25 湖南 山羊 YN2014 2014-04-01 云南 山羊 GD2014 2014-05-15 广东 山羊 SaX2014 2014-04-01 陕西 山羊 SC2014 2014-06-10 四川 山羊 JX2014 2014-04-01 江西 山羊 表 3 PPRV参考毒株信息
Table 3. Details of the PPRV reference strains used in this study
毒株 分离年 来源地 宿主 GenBank号 毒株 分离年 来源地 宿主 GenBank号 Tibet/30/2007 2007 中国西藏 山羊 FJ905304 Ethiopia 2010 2010 埃塞俄比亚 山羊 KJ867541 Tibet/33/2007 2007 中国西藏 山羊 KX421388 Ghana NK1 2010 2010 加纳 山羊 KJ466104 Tibet/Bharal/2008 2008 中国西藏 山羊 JX217850 Oman 1983 1983 阿曼 山羊 KJ867544 Côte d' Ivoire 89 1989 科特迪瓦 山羊 EU267273 UAE 1986 1986 阿联酋 山羊 KJ867545 Nigeria 76/1 1976 尼日利亚 山羊 EU267274 India TN Gingee 2014 2014 印度 山羊 KR261605 Turkey 2000 2000 土耳其 山羊 NC-006383 Uganda 2012 2012 乌干达 山羊 KJ867543 CIV 01 P 2009 2009 科特迪瓦 山羊 KR781451 Morocco 2008 2008 摩洛哥 山羊 KC594074 Ethiopia 1994 1994 埃塞俄比亚 山羊 KJ867540 -
2013−2017年,来自中国21个省份37个疫点的37株PPRV毒株P基因核苷酸序列间遗传距离为0~0.009 2。其中,2013年11月至2014年10月间的18个毒株P基因序列完全一致。这些毒株分别来自新疆(XJYL2013、XJ22013、XJ32013、XJ42013、XJ52013)、甘肃(GS2014)、辽宁(LN2014)、重庆(CQ2014)、云南(YN2014)、江苏(JS2014)、湖北(HB2014)、河南(HeN2014)、山西(SX2014)、吉林(JL2014)、贵州(GZ2014)、湖南(HN2014)、四川(SC2014)、浙江(ZJ82014)等14个省份。毒株XJ302017与GZ252017间的基因核苷酸序列差异最大,为0.009 2,共14个位点存在序列差异。
为考察毒株序列变异与流行时间的关系,选取最早采集的XJYL2013毒株作为参照,将其余36个毒株与其进行P基因核苷酸序列比对。结果发现:36个毒株分别在0~8个位点发生了突变。其中2013年11月至2014年10月间来自14省份的17个毒株与XJYL2013的P基因核苷酸序列完全一致。2014年3月至2015年3月黑龙江的HLJ2014、江西的JX2014、安徽的AH2014、广西的GX2014和广东的GD2014等5个毒株在不同位点发生了1个变异。2014年2月至2016年8月宁夏的NX2014、陕西的SX2014、吉林的JL2014、江西的JX2014和JX62014、浙江的ZJ2014、广西的GX62016等7个毒株在2个位点发生了变异。2015年4月安徽的AH162015和2016年1月的江苏JS142016等2个毒株在3个位点发生了变异。2015年9月贵州的GZ112015、2016年10月湖南的HN182016、2017年3月湖南的HN02017等3个毒株在4个位点发生了变异。2017年9月贵州的GZ252017、2017年2月新疆的XJ302017分别在6和8个位点发生了变异。37个毒株之间的核苷酸变异位点分布于P基因的47个位点,其中45个突变位点仅存在于1个毒株中,2个突变位点分别为3个毒株共有。上述结果表明P基因核苷酸序列突变位点数与毒株流行时间正相关,突变无地域相关性。利用Simplot软件对核苷酸序列发生变异的19株PPRV毒株与XJYL2013进行P基因核苷酸序列同源性比较分析(图1),结果显示:P基因序列的850~1 250 nt间序列同源性较高,突变分布少,500~800 nt区域变异较大。
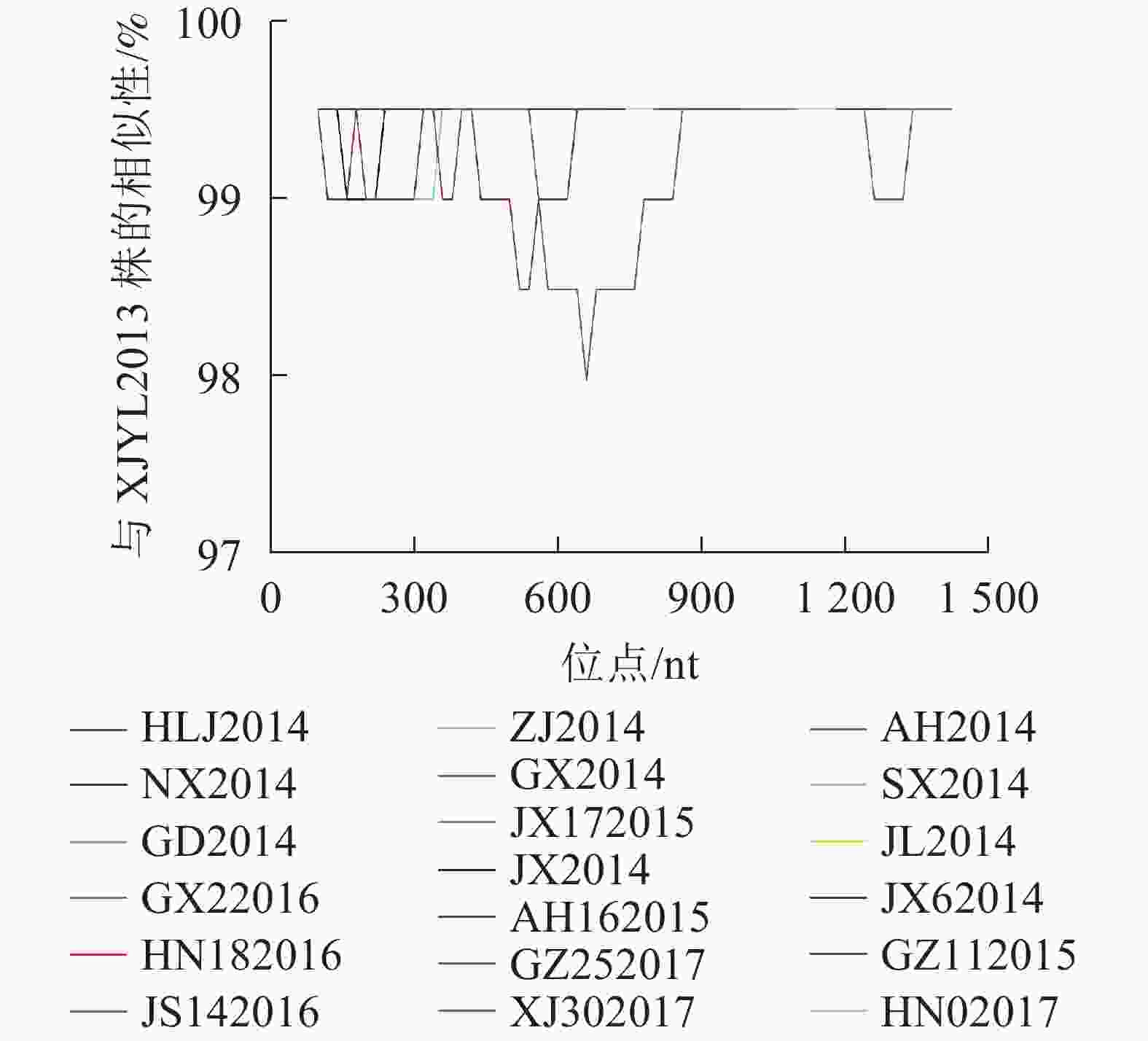
图 1 2013−2017年中国PPRV毒株P基因核苷酸序列同源性Simplot分析
Figure 1. P gene nucleotide sequence similarity of China 2013−2017 PPRV strains using Simplot analysis
P基因的47个核苷酸变异位点中,有26个核苷酸变异导致了氨基酸序列的改变(表4):17个变异发生于第1位密码子,其中的16个位点导致氨基酸改变;9个变异位于第2位密码子,全部导致氨基酸改变;21个变异位于第3位密码子,1个位点导致氨基酸改变。
表 4 2013−2017年中国PPRV毒株P基因核苷酸和氨基酸序列变异情况
Table 4. Nucleotide and amino acid diversity of China 2013−2017 PPRV strains
变异位点 核苷酸/个 氨基酸/个 非同义突变率/% 中国2013−2017年流行毒株 第1位密码子 17 16 94.12 第2位密码子 9 9 100.00 第3位密码子 21 1 4.76 合计 47 26 55.32 中国2013−2017年流行毒株与其他代表毒株 第1位密码子 2 1 50.00 第2位密码子 2 2 100.00 第3位密码子 6 0 0.00 合计 10 3 30.00 -
2013−2017年中国37个PPRV毒株P基因氨基酸序列之间的遗传距离为0~0.007 9。P基因编码的509个氨基酸位点中,25个发生了突变,其中氨基端(1~250位氨基酸)16个,羧基端(251~509位氨基酸) 9个(图2)。第21、83、505位氨基酸位点分别有2个、3个和3个毒株发生了变异。在第21位氨基酸位点,XJ302017第1位密码子发生突变,导致丙氨酸(A)突变为苏氨酸(T),HN182016第2位密码子发生突变,导致丙氨酸(A)突变至缬氨酸(V)。第83位氨基酸位点,3个毒株在同一个核苷酸位点发生不同突变,JL2014和 JX2014导致苯丙氨酸(F)突变至亮氨酸(L),SaX2014苯丙氨酸(F)突变为缬氨酸(V)。第505位氨基酸位点,3个毒株在同一个核苷酸位点发生相同突变,JX62014、HN182016和GZ112015均发生了亮氨酸(L)至缬氨酸(V)的突变。

图 2 2013−2017年中国37个PPRV毒株P蛋白氨基酸序列比对
Figure 2. P protein amino acid sequence alignment of China 2013−2017 PPRV strains
-
比较2013−2017年中国37株PPRV流行毒株与之前15株代表毒株的P基因核苷酸序列,发现遗传距离为0.025 4~0.139 7,其中与2007−2008年中国西藏地区3个毒株P基因核苷酸序列遗传距离为0.025 4~0.031 5,而西藏地区3个流行毒株之间的P基因核苷酸序列遗传距离为0~0.001 3。与15株代表毒株的氨基酸序列比对发现:2013−2017年中国37株PPRV流行毒株P基因的10个位点发生了核苷酸序列突变,其中3个导致了氨基酸序列的改变(表4);2个变异发生于第1位密码子,其中1个导致氨基酸改变;2个变异位于第2位密码子,全部导致氨基酸改变;6个变异位于第3位密码子,未导致氨基酸改变。
2013−2017年,中国37株PPRV流行毒株与15株代表毒株P基因氨基酸序列的3个变异位点均位于175~243 位氨基酸,全部处在P蛋白的氨基端,其中175和176等2个相邻位点都为变异位点。
-
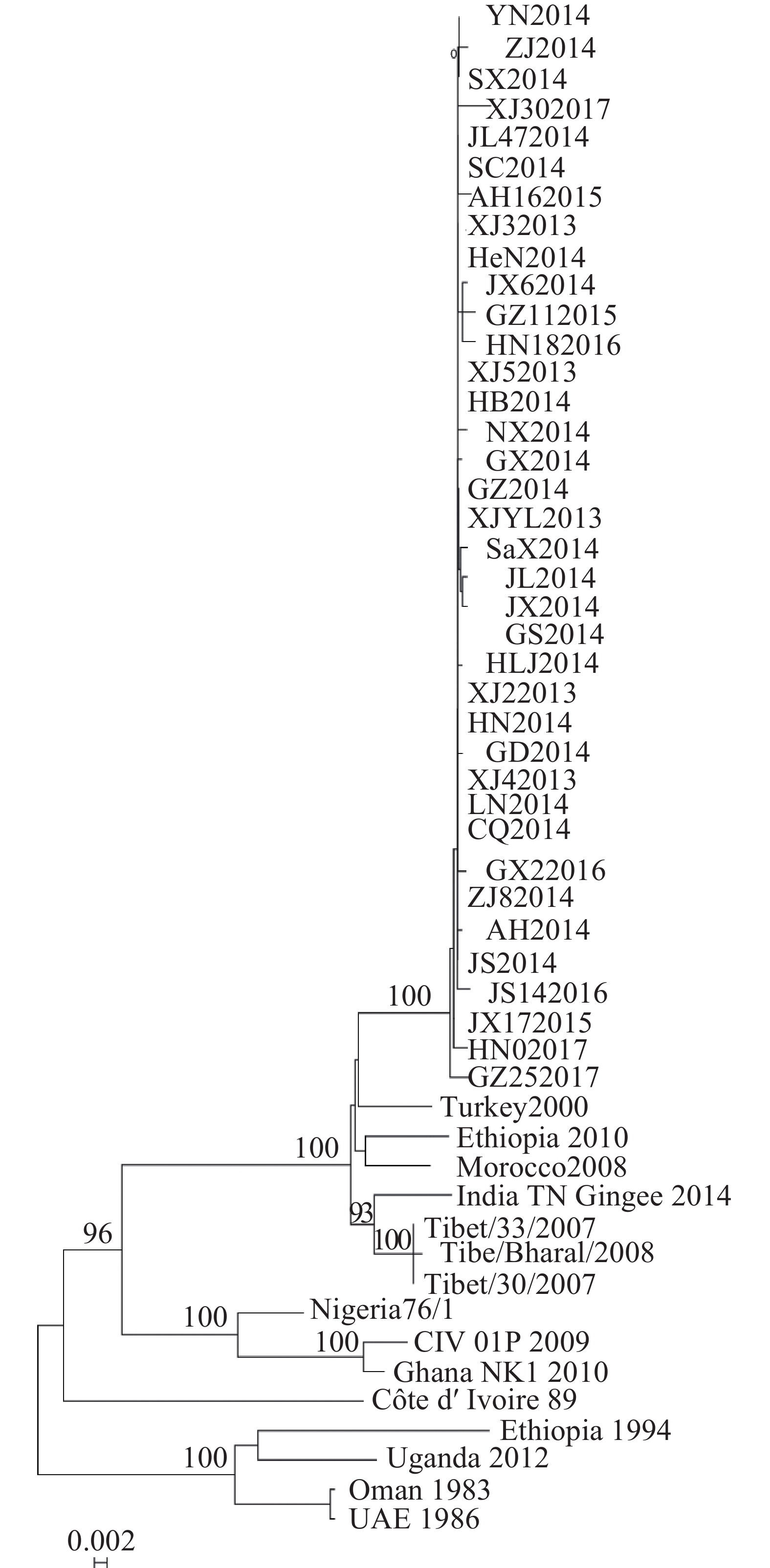
基于P基因核苷酸序列构建系统发育树,发现2013−2017年中国37株PPRV流行毒株构成基因Ⅳ系中1个独立的进化小分支,而中国西藏地区流行毒株与印度2014年毒株形成1个小分支(图3)。
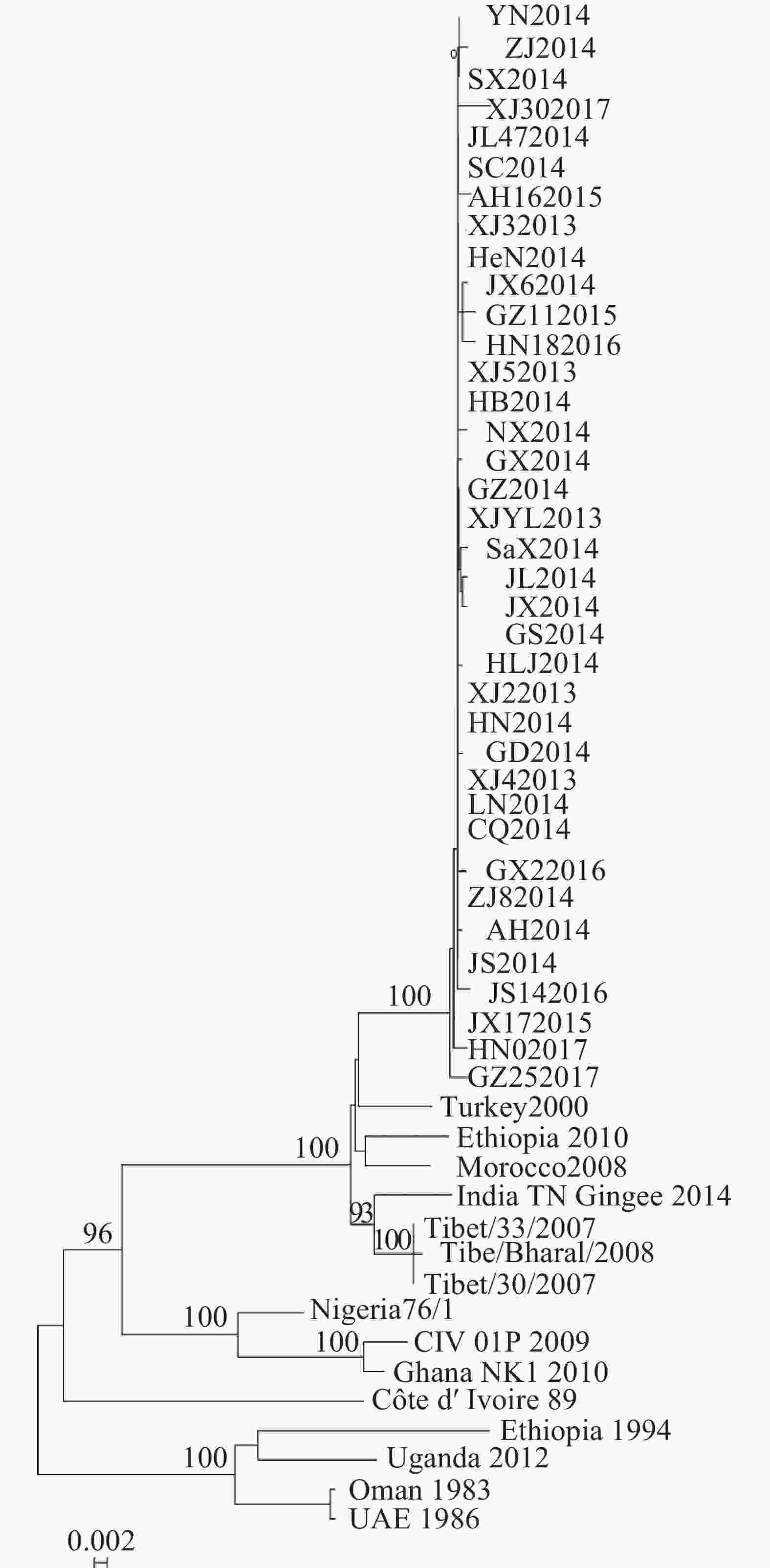
图 3 52个PPRV毒株P基因分子进化树
Figure 3. Phylogenetic relationships of 52 PPRV genome sequences
-
本研究对2013−2017年中国21个省份37个疫点的PPRV流行毒株P基因进行序列分析,探明了PPRV流行过程中P基因序列变异情况,为PPR控制和消灭策略的制定提供了数据支持。
2013−2017年中国37个PPRV流行毒株的P基因变异较大。PPRV在中国流行的4 a间,P基因47个核苷酸位点发生了突变,其中26个导致了氨基酸序列的改变,非同义突变率为55.3%。研究[8]发现:2013年11月至2014年6月,25个毒株P基因在12个位点发生突变,其中10个导致了氨基酸序列的改变,非同义突变率为83.3%,高于F、H、L、N和M基因。已有研究[10]表明:2013−2017年中国37个PPRV流行毒株H基因的非同义突变率为58.5%,高于本研究中的P基因。这一结果提示:实施全面免疫后,PPRV在中国的流行受到了免疫选择压的作用,P基因的非同义突变率降低。
2013−2017年中国流行的PPRV毒株P蛋白氨基酸序列在10个位点发生了特征性的核苷酸突变,其中第21、83和505位3个导致了氨基酸序列改变。已有研究[9]表明:P蛋白对病毒RNA合成至关重要,可以分别与N蛋白、L蛋白及核衣壳结合形成不同的复合体发挥作用。P蛋白与L蛋白结合形成依赖于RNA的RNA聚合酶进行病毒基因组RNA的转录,L蛋白发挥RNA聚合酶(RdRp)的作用,而P蛋白的作用是引导P-L复合体与核衣壳结合。一旦获得足够多的病毒蛋白,N蛋白就与病毒RNA结合,在这个过程中,P蛋白发挥蛋白伴侣的作用,引导N蛋白以游离形式与RNA结合。通常认为[9]:N-P复合体调节从转录到复制的转变。P-L复合体负责病毒基因组的复制,也就是说,先合成全长正链反向基因组RNA,然后以其为模板合成负链基因组RNA,用以组装病毒粒子[11]。P蛋白通过与游离的N蛋白结合,从而阻止N蛋白发生非特异性的自我组装,形成的N0-P复合物在复制过程中对新合成的RNA进行加帽化。副粘病毒P蛋白C末端的X-domain是主要的N蛋白结合位点[9]。PPRV第429~509位氨基酸区域高度保守,推测可能为N蛋白结合位点[12]。P蛋白与其他蛋白相互作用的功能域主要分布于羧基端,因此,在病毒演化过程中,P蛋白的羧基端较为保守。本研究发现:2013−2017年中国流行的PPRV毒株P蛋白氨基酸序列在505位发生了高频突变。该突变对其结构及功能的影响还需要进一步的研究。
在中国流行过程中,PPRV毒株P基因突变位点的数量与毒株流行时间呈正相关。本研究发现:相对于最早分离的XJYL2013,2013−2014年流行的毒株P基因突变位点为0~2个,而2017年流行毒株P基因突变位点为4~8个,随着毒株流行时间增长,P基因突变位点增多。已有研究[8]表明:PPRV毒株基因组在流行过程中的核苷酸突变进化速率为9.54×10−4位点·a−1。在流行过程中PPRV毒株P基因持续发生突变,但不同毒株突变位点不同,表现为相对随机的突变。随着流行时间的增长,在稳定免疫选择压的作用下,是否会出现变异位点的稳定和定植,还有待进一步的研究。持续实时监测PPRV各基因分子变异,对于该病的防控具有重要意义。
Evolution characterization of P gene of peste des petits ruminants virus in China from 2013 to 2017
-
摘要:
目的 探究中国小反刍兽疫病毒(PPRV)田间流行毒株P基因的分子进化特征。 方法 对2013−2017年中国21省份37个疫点的小反刍兽疫病毒流行毒株进行磷蛋白基因(P基因)序列比较和分子进化分析。 结果 37个毒株P基因核苷酸序列间的遗传距离为0~0.009 2,变异分布在47个位点;P蛋白氨基酸序列间的遗传距离为0~0.007 9,变异分布在26个位点。与15株代表毒株的序列比对发现:2013−2017年中国37株PPRV流行毒株P基因的10个位点发生了核苷酸序列突变,其中3个导致了氨基酸序列的改变。以最大似然法构建分子进化树,发现2013−2017年中国流行的37个毒株构成基因Ⅳ系中1个独立的进化分支,与2007年传入西藏的毒株处于不同的进化分支。 结论 2013−2017年中国小反刍兽疫病毒P基因变异较大;随毒株流行时间增长,P基因的突变位点增多,但不同毒株突变位点不同,表现为相对随机的突变。图3表4参12 Abstract:Objective The objective of this study is to explore the molecular evolution characteristics of P gene of peste des petits ruminants virus (PPRV) during an epidemic in China. Method Bioinformatics analysis on the sequences of P gene of PPRV strains from 37 epidemic sites in 21 provinces of China from 2013 to 2017 was carried out. Result The genetic distances among the nucleotide sequences of P gene of 37 strains was 0−0.0092, and the variation sites were distributed in 47 sites. The genetic distances among the amino acid sequences of P gene of these strains ranged from 0−0.0079, and the variation sites were distributed in 26 sites. Sequence comparison with 15 representative strains showed that nucleotide sequence mutations occurred at 10 sites of P gene of 37 PPRV epidemic strains in China since 2013, and 3 of them led to the change of amino acid sequence. Phylogenetic analysis of P gene using maximum likelihood method revealed that all the 37 PPRV strains prevalent in China from 2013 to 2017 could be grouped into an independent clade in lineageⅣ, different from the strain introduced into Tibet in 2007. Conclusion From 2013 to 2017, the P gene mutation of peste des petits ruminants virus in China was relatively large. With the increase of epidemic time, the mutation sites of P gene increased, but the mutation sites of different strains were different, showing a relatively random mutation. [Ch, 3 fig. 4 tab. 12 ref.] -
Key words:
- peste des petits ruminants virus /
- P gene /
- sequence mutation /
- genetic evolution
-
图 1 2013−2017年中国PPRV毒株P基因核苷酸序列同源性Simplot分析
Figure 1 P gene nucleotide sequence similarity of China 2013−2017 PPRV strains using Simplot analysis
图 2 2013−2017年中国37个PPRV毒株P蛋白氨基酸序列比对
Figure 2 P protein amino acid sequence alignment of China 2013−2017 PPRV strains
表 1 2014−2017年中国12株PPRV毒株信息
Table 1. Details of the 12 PPRV strains collected in China during 2014−2017
毒株编号 采样时间(年-月-日) 来源省份 毒株编号 采样时间(年-月-日) 来源省份 JL472014 2014-04-06 吉林 JS142016 2016-01-20 江苏 JX62014 2014-07-21 江西 GX22016 2016-08-22 广西 ZJ82014 2014-10-11 浙江 HN182016 2016-10-16 湖南 JX172015 2015-03-30 江西 XJ302017 2017-02-12 新疆 AH162015 2015-04-09 安徽 HN02017 2017-03-05 湖南 GZ112015 2015-09-02 贵州 GZ252017 2017-09-21 贵州  下载: 导出CSV
下载: 导出CSV
表 2 2013−2014年中国PPRV流行毒株信息
Table 2. Details of the PPRV strains collected in China during 2013−2014
病毒名称 采样时间(年-月-日) 来源省份 宿主 病毒名称 采样时间(年-月-日) 来源省份 宿主 XJYL2013 2013-11-30 新疆 山羊 JL2014 2014-04-01 吉林 绵羊 XJ22013 2013-12-20 新疆 山羊 JS2014 2014-04-02 江苏 山羊 XJ32013 2013-12-21 新疆 绵羊 HeN2014 2014-04-03 河南 山羊 XJ42013 2013-12-22 新疆 山羊 HB2014 2014-04-03 湖北 山羊 XJ52013 2013-12-29 新疆 山羊 AH2014 2014-04-03 安徽 山羊 GS2014 2014-01-22 甘肃 绵羊 SX2014 2014-04-05 山西 山羊 NX2014 2014-02-17 宁夏 绵羊 GX2014 2014-04-16 广西 山羊 LN2014 2014-03-17 辽宁 山羊 GZ2014 2014-04-21 贵州 山羊 CQ2014 2014-03-30 重庆 山羊 ZJ2014 2014-04-25 浙江 山羊 HLJ2014 2014-03-31 黑龙江 山羊 HN2014 2014-04-25 湖南 山羊 YN2014 2014-04-01 云南 山羊 GD2014 2014-05-15 广东 山羊 SaX2014 2014-04-01 陕西 山羊 SC2014 2014-06-10 四川 山羊 JX2014 2014-04-01 江西 山羊
下载: 导出CSV
表 3 PPRV参考毒株信息
Table 3. Details of the PPRV reference strains used in this study
毒株 分离年 来源地 宿主 GenBank号 毒株 分离年 来源地 宿主 GenBank号 Tibet/30/2007 2007 中国西藏 山羊 FJ905304 Ethiopia 2010 2010 埃塞俄比亚 山羊 KJ867541 Tibet/33/2007 2007 中国西藏 山羊 KX421388 Ghana NK1 2010 2010 加纳 山羊 KJ466104 Tibet/Bharal/2008 2008 中国西藏 山羊 JX217850 Oman 1983 1983 阿曼 山羊 KJ867544 Côte d' Ivoire 89 1989 科特迪瓦 山羊 EU267273 UAE 1986 1986 阿联酋 山羊 KJ867545 Nigeria 76/1 1976 尼日利亚 山羊 EU267274 India TN Gingee 2014 2014 印度 山羊 KR261605 Turkey 2000 2000 土耳其 山羊 NC-006383 Uganda 2012 2012 乌干达 山羊 KJ867543 CIV 01 P 2009 2009 科特迪瓦 山羊 KR781451 Morocco 2008 2008 摩洛哥 山羊 KC594074 Ethiopia 1994 1994 埃塞俄比亚 山羊 KJ867540
下载: 导出CSV
表 4 2013−2017年中国PPRV毒株P基因核苷酸和氨基酸序列变异情况
Table 4. Nucleotide and amino acid diversity of China 2013−2017 PPRV strains
变异位点 核苷酸/个 氨基酸/个 非同义突变率/% 中国2013−2017年流行毒株 第1位密码子 17 16 94.12 第2位密码子 9 9 100.00 第3位密码子 21 1 4.76 合计 47 26 55.32 中国2013−2017年流行毒株与其他代表毒株 第1位密码子 2 1 50.00 第2位密码子 2 2 100.00 第3位密码子 6 0 0.00 合计 10 3 30.00
下载: 导出CSV
-
[1] WANG Zhiliang, BAO Jingyue, WU Xiaodong, et al. Peste des petits ruminants virus in Tibet, China [J]. Emerging Infect Dis, 2009, 15(2): 299 − 301. [2] BAO Jingyue, WANG Zhiliang, LI Lin, et al. Detection and genetic characterization of peste des petits ruminants virus in free-living bharals (Pseudois nayaur) in Tibet, China [J]. Res Vet Sci, 2011, 90(2): 238 − 240. [3] WU X, LI Linde, LI Jinming, et al. Peste des petits ruminants viruses re-emerging in China, 2013−2014 [J]. Transboundary Emerging Dis, 2016, 63(5): e441 − 446. doi: 10.1111/tbed.12308. [4] 王清华, 刘春菊, 吴晓东, 等. 新疆小反刍兽疫疫情诊断[J]. 中国动物检疫, 2014, 31(1): 72 − 75. WANG Qinghua, LIU Chunju, WU Xiaodong, et al. Diagnosis of peste des petis ruminants in goats in Xinjiang, China [J]. China Anim Health Insp, 2014, 31(1): 72 − 75. [5] KWIATEK O, MINET C, GRILLET C, et al. Peste des petits ruminants (PPR) outbreak in Tajikistan [J]. J Comp Pathol, 2007, 136(2/3): 111 − 119. [6] SHAILA M S, SHAMAKI D, FORSYTH M A, et al. Geographic distribution and epidemiology of peste des petits ruminants viruses [J]. Virus Res, 1996, 43(2): 149 − 153. [7] MUNIRAJU M, MUNIR M, PARTHIBAN A B R, et al. Molecular evolution of peste des petits ruminants virus [J]. Emerging Infect Dis, 2014, 20(12): 2023 − 2033. [8] BAO Jingyue, WANG Qinghua, LI Lin, et al. Evolutionary dynamics of recent peste des petits ruminants virus epidemic in China during 2013−2014 [J]. Virology, 2017, 510: 156 − 164. [9] SHAJI D, SHAILA M S. Domains of rinderpest virus phosphoprotein involved in interaction with itself and the nucleocapsid protein [J]. Virology, 1999, 258(2): 415 − 424. [10] 刘春菊, 盛琰翔, 王清华, 等. 2013−2017年我国小反刍兽疫病毒H基因分子演化特征[J]. 中国动物检疫, 2019, 36(9): 19 − 24. LIU Chunju, SHENG Yanxiang, WANG Qinghua, et al. Molecular evolution characterization of H gene of peste des petits ruminants virus in China during 2013 to 2017 [J]. China Anim Health Insp, 2019, 36(9): 19 − 24. [11] KARLIN D, FERRON F, CANARD B, et al. Structural disorder and modular organization in Paramyxovirinae N and P [J]. J Gen Virol, 2003, 84(12): 3239 − 3252. [12] 包静月, 赵文姬, 李林, 等. 中国西藏小反刍兽疫病毒China/Tib/Gei/07-30磷蛋白基因的分子特征及其编码蛋白特性分 析[J]. 病毒学报, 2011, 27(1): 26 − 33. BAO Jingyue, ZHAO Wenji, LI Lin, et al. Sequence analysis of the phosphoprotein gene of peste des petits ruminants virus of Chinese origin [J]. Chin J Virol, 2011, 27(1): 26 − 33. -
-
链接本文:
https://zlxb.zafu.edu.cn/article/doi/10.11833/j.issn.2095-0756.20190742
 点击查看大图
点击查看大图

计量
- 文章访问数: 2287
- HTML全文浏览量: 581
- PDF下载量: 30
- 被引次数: 0
