





-
叶绿体是细胞中具有独立基因组的细胞器,其结构简单、分子量相对较小、进化速率较慢、具有良好的保守性,在物种鉴定和系统发育分析中得到了广泛的应用[1]。在大多数植物中,叶绿体基因组的突出特征是存在1个大的倒置重复序列区(IR),IR被1个大单拷贝区(LSC)和1个小单拷贝区(SSC)隔开,叶绿体DNA大小变化大多可以通过IR区域长度的变化来解释[2−3]。目前,叶绿体基因组已广泛应用于苔藓植物的系统发育研究中[4−5]。与核基因组和线粒体基因组相比,叶绿体基因组结构简单且保守,全序列易获得,相对于多片段研究也有更好的解析[6−7]。
新绒苔Neotrichocolea bissetii隶属于新绒苔属 Neotrichocolea,该属仅包含新绒苔1种,为东亚特有属。新绒苔植物体绒毛状,深绿色或黄绿色,有光泽;茎匍匐或倾斜,3至4回羽状分枝,长达10 cm;通常生长在潮湿的石头上或林下溪边的潮湿土壤上,偶见于腐木;主要分布在中国、日本[8]和朝鲜半岛[9]。依据中国苔藓植物濒危等级的评估原则,该种在整个分布区范围内种群变化不详,在黄山已被证实种群数量急剧减少,目前被列为易危物种(VU)[10],该种也被《世界自然保护联盟濒危物种红色名录》列为易危物种。
早期基于形态学的研究,新绒苔被分别放在毛叶苔科Ptilidiaceae[11]或新绒苔科Neotrichocoleaceae[12]。LIU等[13]基于基因片段分析,深入研究了新绒苔属和囊绒苔属Trichocoleopsis的系统发育关系及分类位置,研究结果支持新绒苔放置在新绒苔科,但需要注意的是,其系统进化树中的分支支持率并不高。最新的苔藓植物系统发育研究中均未包含新绒苔的叶绿体基因组数据。鉴于叶绿体基因组信息更系统全面,本研究将基于叶绿体基因组数据,进一步明确新绒苔的系统位置。
本研究利用新一代测序技术(NGS)对新绒苔进行测序,通过组装和注释获得完整的叶绿体基因组序列,分析了新绒苔基因组的序列特征,并结合17种已发表的苔藓植物序列构建了系统发育树,明确了新绒苔的系统位置,为新绒苔的物种鉴定、资源保护和系统进化研究提供了科学依据。
-
新绒苔标本采集于安徽黄山(30°14′N,118°16′E),凭证标本(20220715-76A)经硅胶干燥后保存于安徽师范大学生命科学学院植物标本馆。使用改良十六烷基三甲基溴化铵(CTAB)法提取和纯化叶片组织的总DNA[14]。通过琼脂糖凝胶电泳和分光光度法测定DNA产物的质量和浓度。所有合格的DNA样本送往中国西南野生生物种质资源库分子生物学实验中心进行建库和测序。
-
使用SOAPnuke Toolkit v.1.3.0[15]过滤原始序列(raw reads)获得高质量序列(clean reads)用于后续分析。通过GetOrganelle v1.7[16]组装叶绿体基因组。将组装结果导入Bandage v.8.1[17]中可视化叶绿体基因组结构,去除低覆盖率的低质量片段,得到单倍体叶绿体的全基因组环结构。使用在线工具CPGAVAS2进行注释[18]。用在线程序Organellar Genome DRAW绘制叶绿体的全基因组物理图谱[19]。
-
使用在线程序MIcroSAtellite对新绒苔叶绿体中的简单重复序列(SSR)进行分析[20]。采用CodonW软件[21]分析密码子的使用情况。使用REPuter在线网站[22]分析叶绿体基因组序列,共检测到4种类型的长重复序列:正向重复、反向重复、回文重复和互补重复。
-
利用新测序的新绒苔、囊绒苔T. sacculata与美国国家生物技术信息中心(NCBI)数据库中公布的5种苔藓植物(登录号分别为NC_010359、NC_019628、NC_019628、NC_043786、NC_043789)的叶绿体基因组序列进行比较,分析叶绿体基因组的总基因数、GC含量、LSC、SSC和IR。利用在线工具IRscope可视化叶绿体基因组的LSC/IR/SSC边界变化[23]。
-
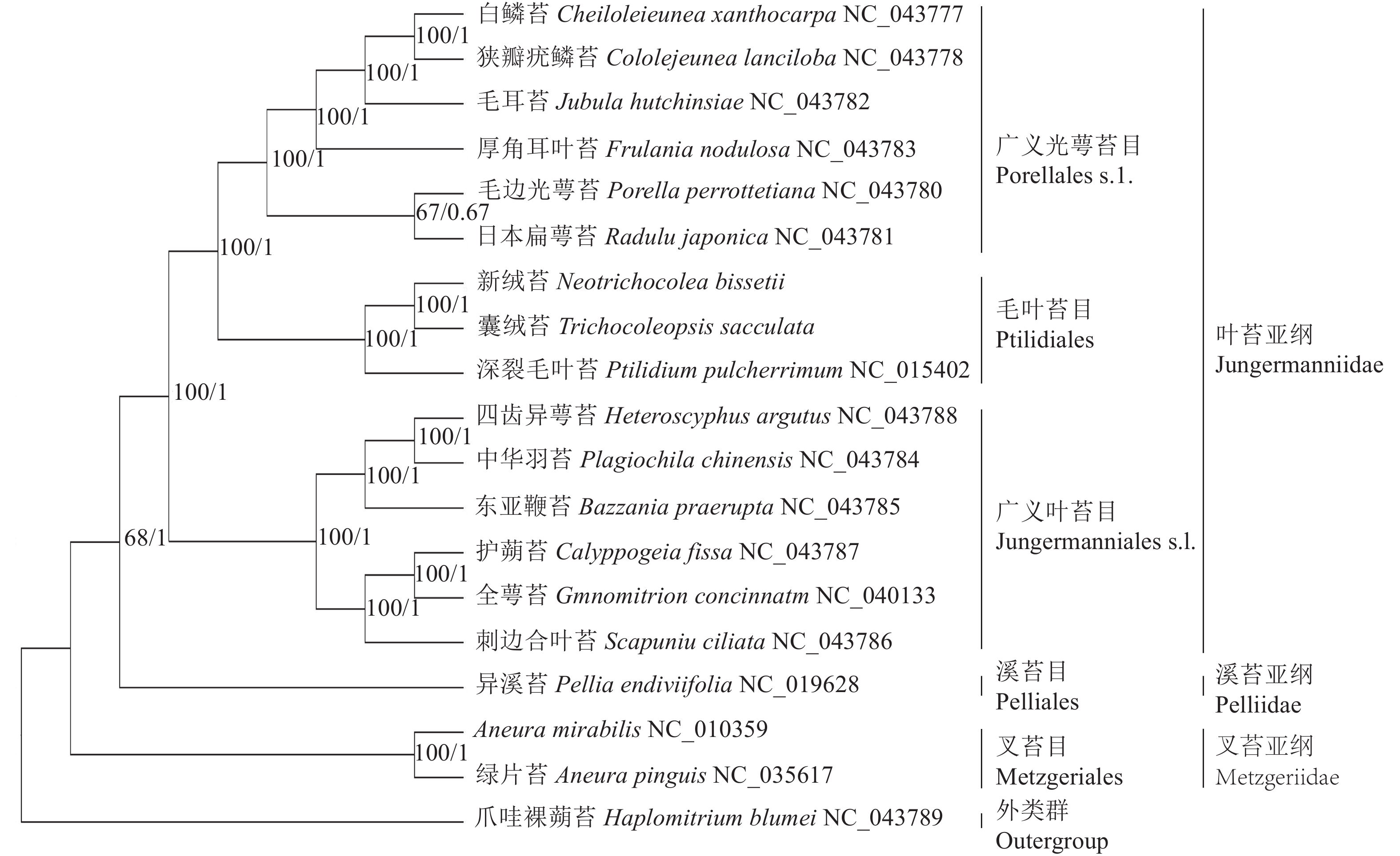
基于苔藓植物系统发育纲要[4]和YU等[5]的研究,以爪哇裸蒴苔Haplomitrium blumei (NC_043789)为外类群,基于新绒苔、囊绒苔和NCBI数据库下载已公布的17种苔藓植物(表1)的叶绿体基因组序列构建系统发育树。这17种苔藓植物均属于叶苔纲Jungermanniopsida,包含了其下的3个亚纲代表类群:叉苔亚纲Metzgeriidae的绿片苔Aneura pinguis和A. mirabilis,溪苔亚纲Pelliidae的异溪苔Pellia endiviifolia,其余14种为叶苔亚纲Jungermanniidae。
物种 GenBank 登录号 物种 GenBank 登录号 Aneura mirabilis NC_010359 四齿异萼苔Heteroscyphus argutus NC_043788 绿片苔Aneura pinguis NC_035617 毛耳苔Jubula hutchinsiae NC_043782 东亚鞭苔Bazzania praerupta NC_043785 异溪苔Pellia endiviifolia NC_019628 护蒴苔Calypogeia fissa NC_043757 中华羽苔Plagiochila chinensis NC_043784 白鳞苔Cheilolejeunea xanthocarpa NC_043777 毛边光萼苔Porella perrottetiana NC_043780 狭瓣疣鳞苔Cololejeunea lanciloba NC_043778 深裂毛叶苔Ptilidium pulcherrimum NC_015402 厚角耳叶苔Frullania nodulosa NC_043783 日本扁萼苔Radula japonica NC_043781 全萼苔Gymnomitrion concinnatum NC_040133 刺边合叶苔Scapania ciliata NC_043786 爪哇裸蒴苔Haplomitrium blumei NC_043789 Table 1. Information of 17 samples of bryophytes
利用MAFFT程序对新绒苔和其他苔藓植物的叶绿体基因组进行全基因组比较[24−25]。使用PhyloSuite v.1.2.2[26]的ModelFinder插件分别计算最大似然(ML)和贝叶斯推断(BI)树模型。ML树和BI树分别使用RAxML v.7.2.8[27]和MrBayes v.3.1.2[28]构建。
-
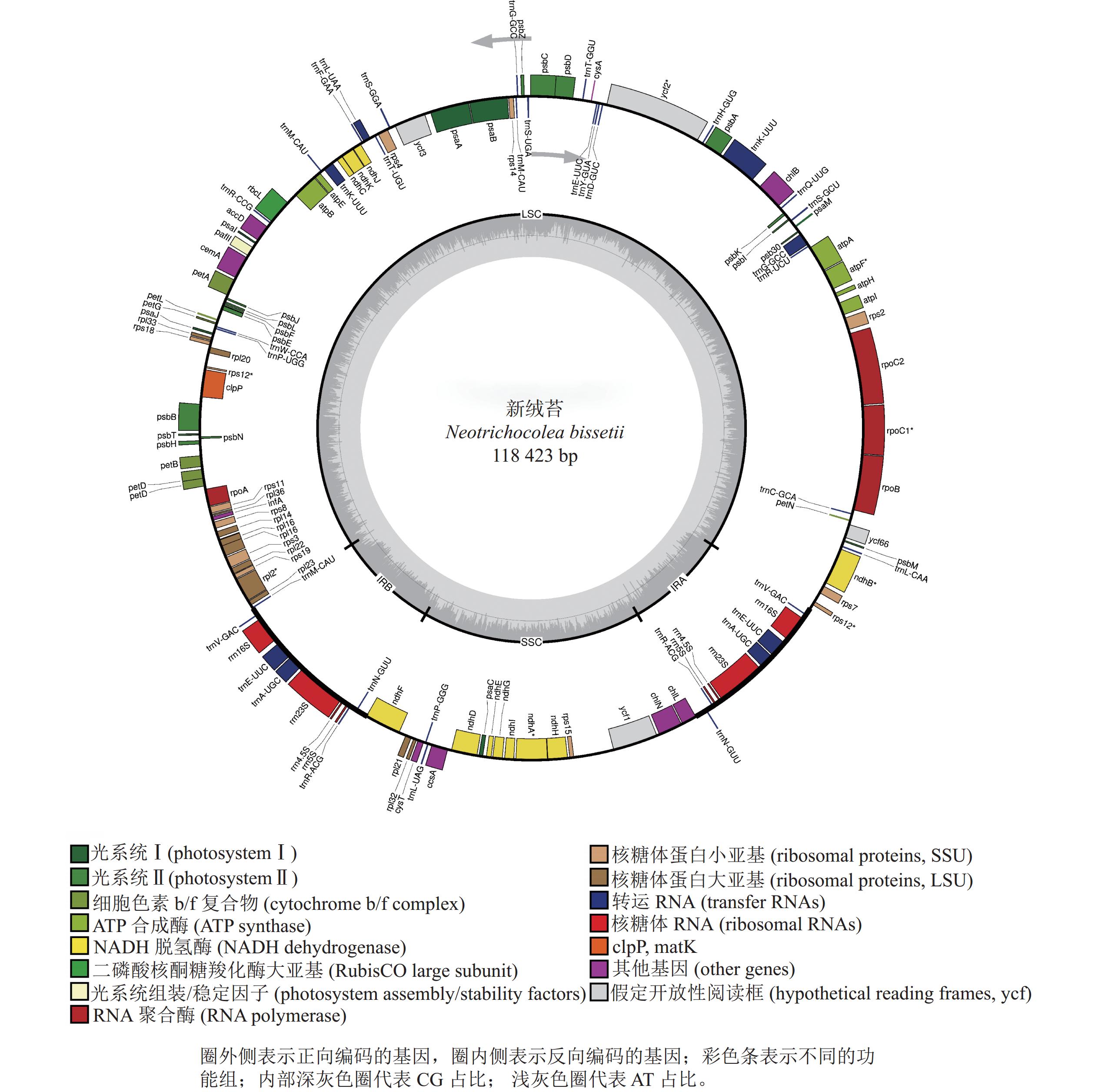
新绒苔叶绿体基因组为典型的四分体结构,总长度为118 423 bp,其中包括1对IR (9 031 bp)、1个LSC (80 837 bp)和1个SSC (19 524 bp)(图1)。全基因组GC含量为33.3%,IR的GC含量(47.0%)高于SSC (31.2%)和LSC (29.3%)。新绒苔叶绿体基因组序列共编码132个基因,其中蛋白编码基因87个,rRNA基因8个,tRNA基因37个。
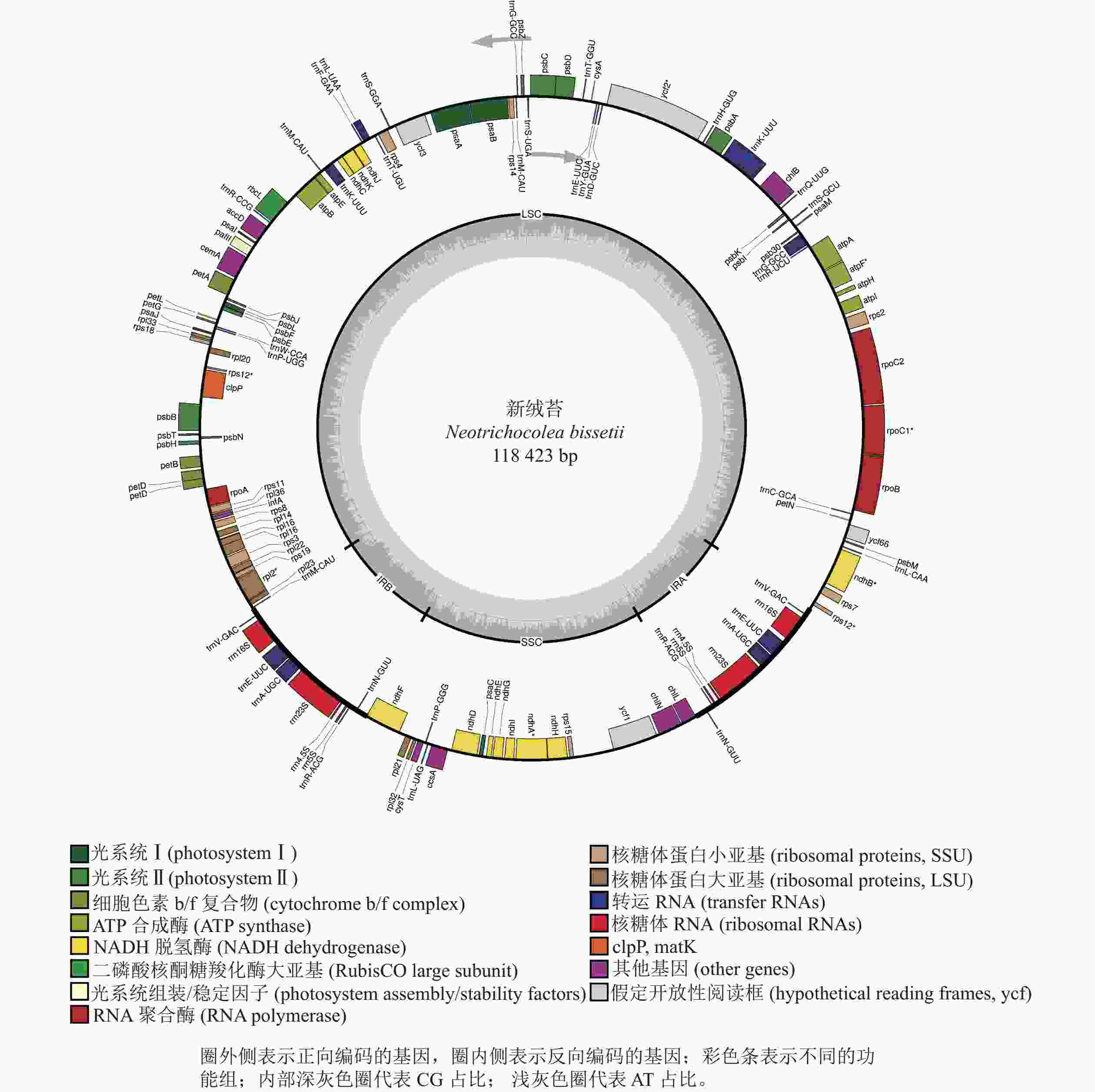
Figure 1. Chloroplast genome gene map of N. bissetii
按照基因功能,主要分为光合作用、转录和未知功能基因,未发现转录起始因子infA。叶绿体基因内含子分析显示:10个基因含有内含子,clpP蛋白编码基因含有2个内含子(表2)。
基因类别 基因功能 基因名称 光合作用 ATP合酶亚基 atpA, atpB, atpE, atpH, atpI 光系统Ⅰ psaA, psaB, psaC, psaJ 光系统Ⅱ psbA, psbB, psbC, psbD, psbE, psbL, psbN, psbT, psbZ, ycf3 NADH-脱氢酶亚基 ndhA*, ndhB*, ndhC, ndhD, ndhE, ndhF, ndhH, ndhI, ndhJ, ndhK 细胞色素复合物b/f亚基 petA, petB *, petD, petG Rubisco酶亚基 rbcL 复制基因 核糖体大亚基 rpl14, rpl16, rpl2*, rpl20, rpl22, rpl33, rpl36 DNA依赖核酸聚合酶 rpoA, rpoB, rpoC1*, rpoC2 核糖体小亚基 rps12*, rps19, rps2, rps3, rps4, rps8 核糖体RNA基因 rrn16S (×2), rrn23S (×2), rrn4.5S (×2), rrn5S (×2) 转运RNA基因 trnA-UGC (×2)*, trnC-GCA, trnD-GUC, trnE-UUC, trnE-UUC (×2)*, trnF-GAA, trnG-GCC (×2)*, trnH-GUG, trnK-UUU (×2)*, trnL-CAA, trnL-UAA*, trnL-UAG, trnM-CAU (×2), trnN-GUU (×2), trnP-UGG, trnQ-UUG, trnR-ACG (×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-CGU*, trnT-GGU, trnT-UGU, trnV-GAC (×2), trnW-CCA, trnY-GUA 其他基因 乙酰辅酶A羧化酶亚基
c型细胞色素合成基因accD
ccsA蛋白酶
包裹膜蛋白
成熟酶clpP**
cemA
matK保守开放性阅读框 ycf1,ycf2 (×2),ycf66 说明:*和**标记的分别为含有单内含子和双内含子的基因。位于IR的重复基因标记为(×2)。 Table 2. Genes encoded by the chloroplast genome of N. bissetii
-
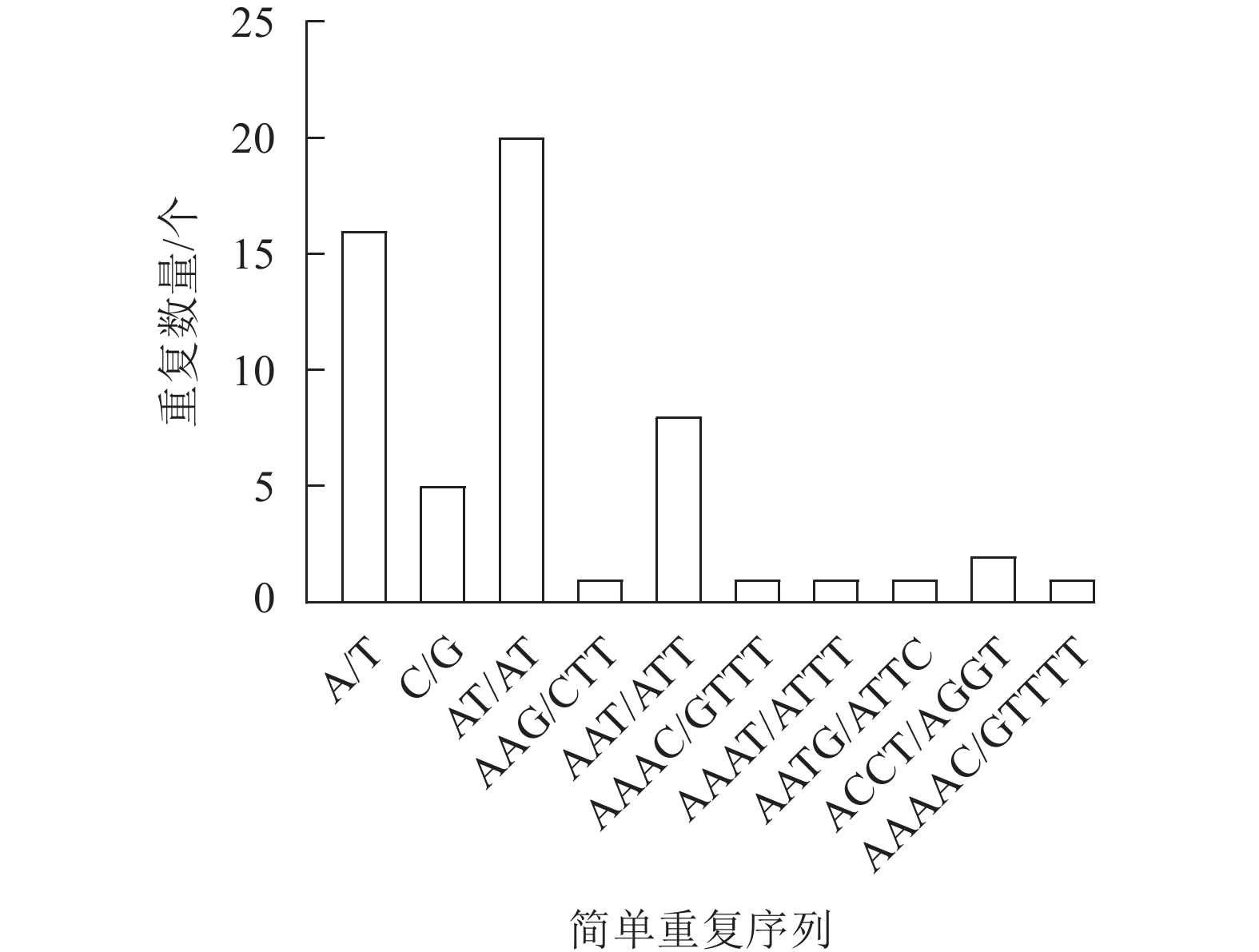
新绒苔叶绿体基因组共检测到56个SSR,包括21个单核苷酸重复序列、20个二核苷酸重复序列、9个三核苷酸重复序列、5个四核苷酸重复序列和1个五核苷酸重复序列。SSR最丰富的类型是单核苷酸重复序列,单核苷酸重复序列以A和T碱基为主。此外,A/T、AT/AT和AAAT/ATTT重复序列占66.07%,说明新绒苔的SSR偏好使用A和T碱基(图2)。
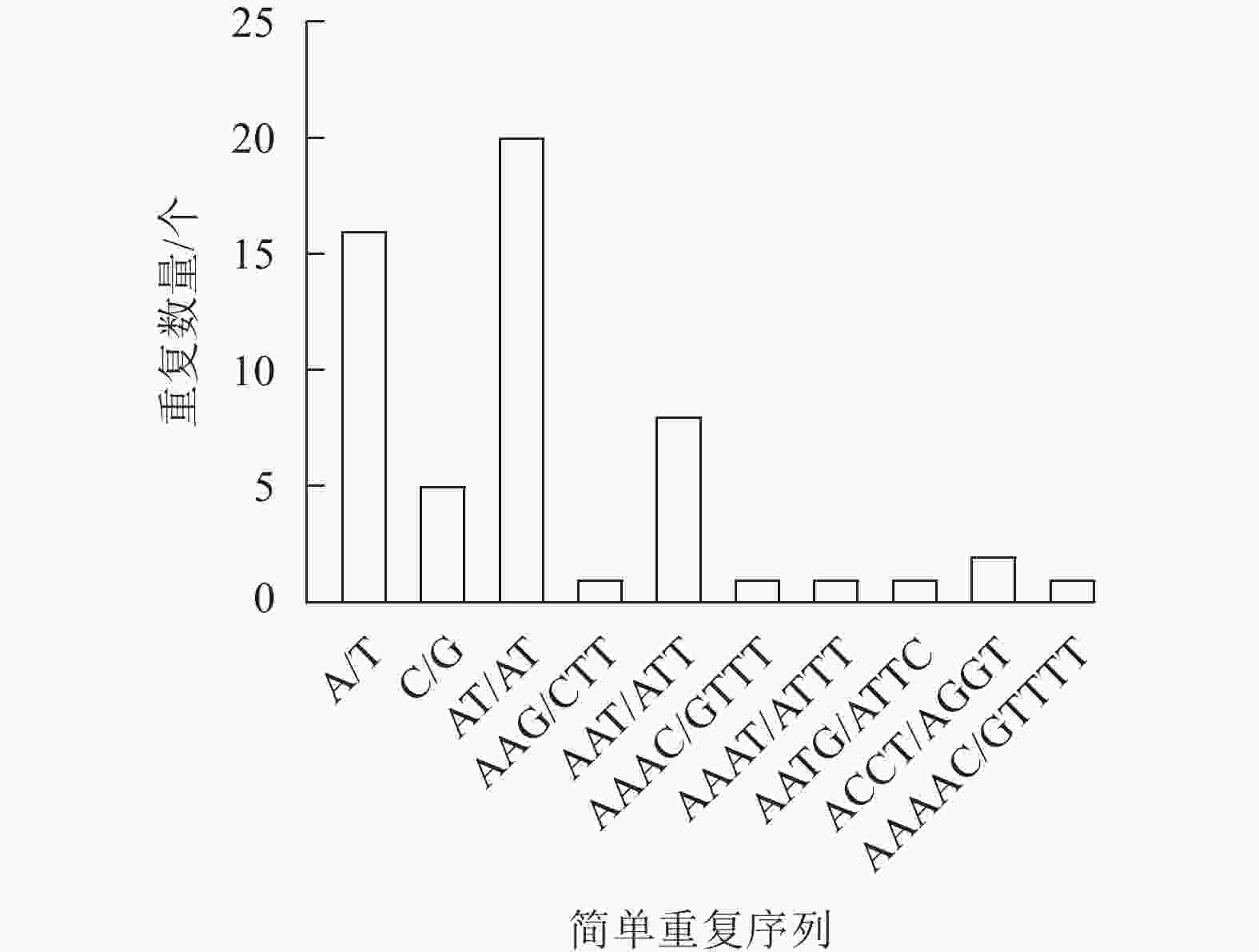
Figure 2. SSR information in chloroplast genome of N. bissetii
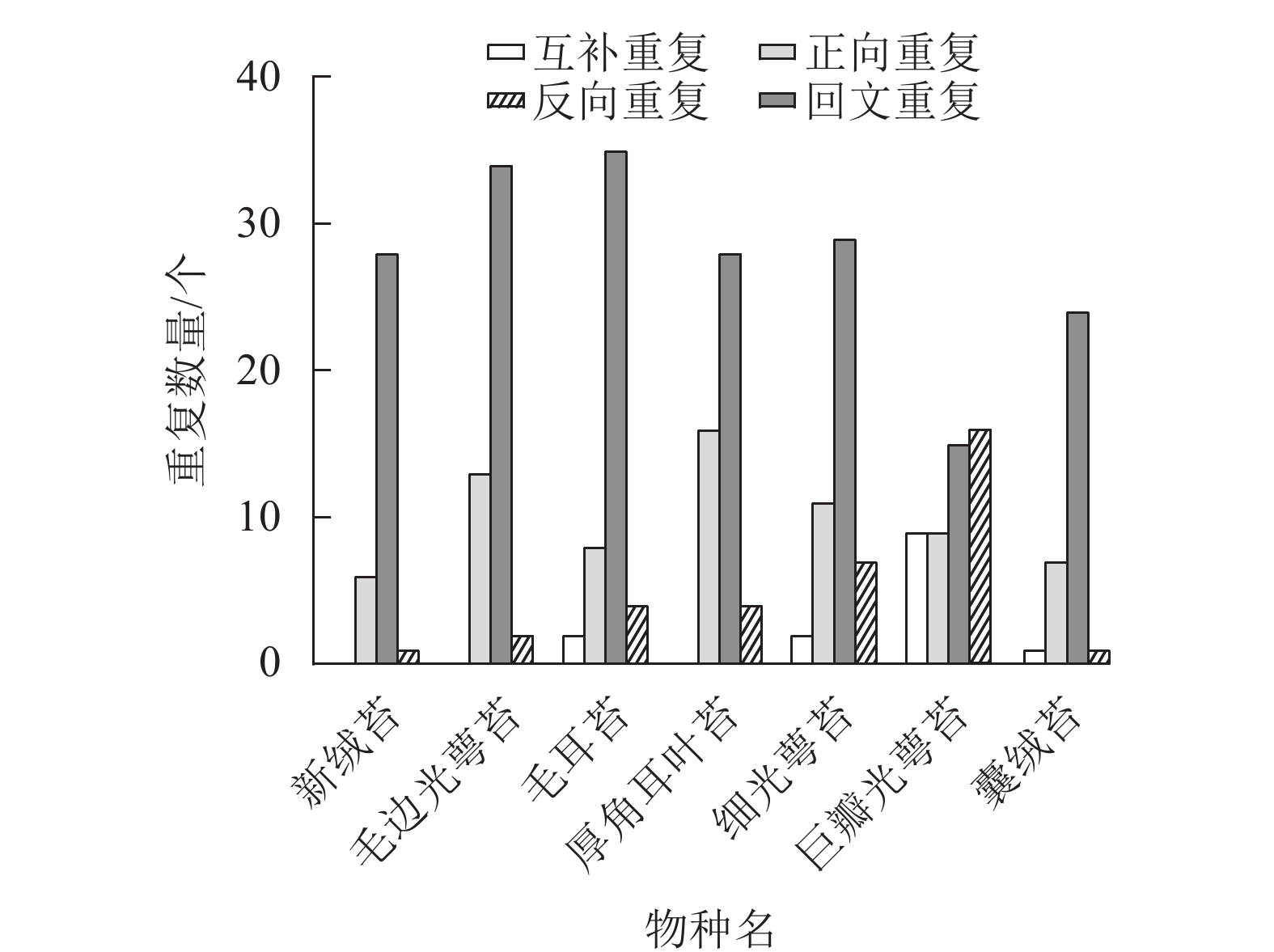
通过分析新绒苔的长重复序列,在叶绿体基因组中鉴定出35个重复序列,其中正向重复6个,回文重复28个,反向重复1个。与NCBI数据库中的毛边光萼苔、毛耳苔、厚角耳叶苔、细光萼苔Porella gracillima (NC_082430)、巨瓣光萼苔Porella grandiloba (NC_072065)在重复数和重复类型上存在差异。新绒苔、毛边光萼苔和厚角耳叶苔未检测到互补重复序列,其余均含有4个重复序列。除巨瓣光萼苔外,其他6种植物重复序列中,回文重复比例最高(图3)。
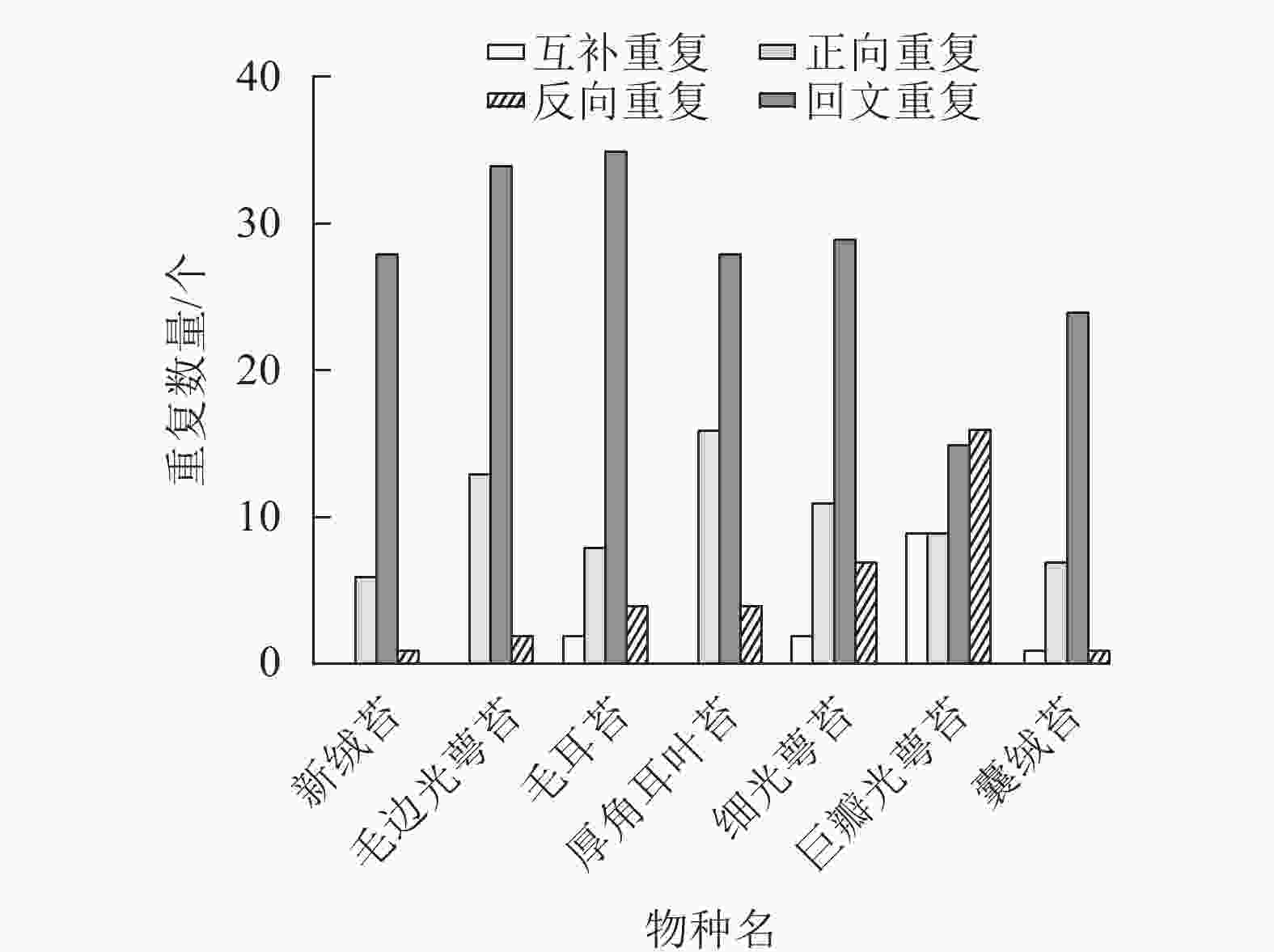
Figure 3. Long repeats of chloroplast genome in 7 bryophytes
-
新绒苔叶绿体基因组中共发现39 474个编码密码子,其中编码亮氨酸的密码子最多,共有4 136个;编码色氨酸的密码子最少,共有475个。64种密码子共编码20种氨基酸,终止密码子为UAA、UAG和UGA。对该叶绿体基因组的相对同义密码子使用度(RSCU)进行分析,RSCU≥1.00的密码子有33个,以A/U结尾的高频密码子28个,以C/G结尾的密码子5个。此外,新绒苔叶绿体基因组偏好密码子多以A/U结尾,与GC含量较低的基因组一致(表3)。
氨基酸 密码子 数量 RSCU 氨基酸 密码子 数量 RSCU 氨基酸 密码子 数量 RSCU 氨基酸 密码子 数量 RSCU Phe UUU 2166 1.36 Tyr UAU 1379 1.42 Glu GAG 411 0.62 Ala GCC 177 0.69 Phe UUC 1027 0.64 Tyr UAC 566 0.58 Ser UCU 637 1.21 Ala GCA 307 1.20 Leu UUA 1196 1.74 Ter UAA 1299 1.47 Ser UCC 496 0.94 Ala GCG 167 0.65 Leu UUG 834 1.21 Ter UAG 608 0.69 Ser UCA 650 1.23 Cys UGU 470 1.14 Leu CUU 746 1.08 Ter UGA 739 0.84 Ser UCG 443 0.84 Cys UGC 353 0.86 Leu CUC 422 0.61 His CAU 644 1.40 Ser AGU 495 0.94 Trp UGG 475 1.00 Leu CUA 571 0.83 His CAC 277 0.60 Ser AGC 437 0.83 Arg CGU 269 0.75 Leu CUG 367 0.53 Gln CAA 799 1.32 Pro CCU 383 1.12 Arg CGC 146 0.41 Ile AUU 1626 1.36 Gln CAG 410 0.68 Pro CCC 314 0.92 Arg CGA 395 1.11 Ile AUC 686 0.58 Asn AAU 1627 1.42 Pro CCA 470 1.38 Arg CGG 204 0.57 Ile AUA 1264 1.06 Asn AAC 658 0.58 Pro CCG 196 0.58 Arg AGA 720 2.02 Met AUG 609 1.00 Lys AAA 2099 1.46 Thr ACU 490 1.16 Arg AGG 404 1.13 Val GUU 626 1.45 Lys AAG 782 0.54 Thr ACC 425 1.01 Gly GGU 411 1.13 Val GUC 250 0.58 Asp GAU 641 1.43 Thr ACA 475 1.13 Gly GGC 207 0.57 Val GUA 558 1.30 Asp GAC 257 0.57 Thr ACG 293 0.70 Gly GGA 526 1.44 Val GUG 288 0.67 Glu GAA 917 1.38 Ala GCU 374 1.46 Gly GGG 316 0.87 Table 3. Analysis of chloroplast genome codon usage in N. bissetii
-
对新绒苔等7种苔藓植物叶绿体基因组的IR边界收缩扩张进行分析(图4),结果表明:7种苔藓植物的叶绿体基因组长度从Aneura mirabilis的108 007 bp到爪哇裸蒴苔的128 728 bp不等。IR长度为16 480~21 856 bp,LSC长度为77 553~88 291 bp,SSC长度为13 974~19 854 bp。trnV、trnL、ndhF、16S、chlL是位于IR边界附近的主要基因。在这7个物种中,rps12、rpl23和trnL基因均位于LSC,trnV基因位于IRa。除爪哇裸蒴苔外,其余物种LSC与IRb之间的连接位点(JSB)的边界基因均为ndhF。新绒苔、囊绒苔、A. mirabilis、异溪苔、爪哇裸蒴苔在叶绿体基因组中IRa与LSC之间的连接位点(JLA)边界未注释到16S基因。新绒苔和囊绒苔在叶绿体基因组中LSC与IRb之间的连接位点(JLB)边界附近缺失trnL基因,明显有别于其他5个物种。
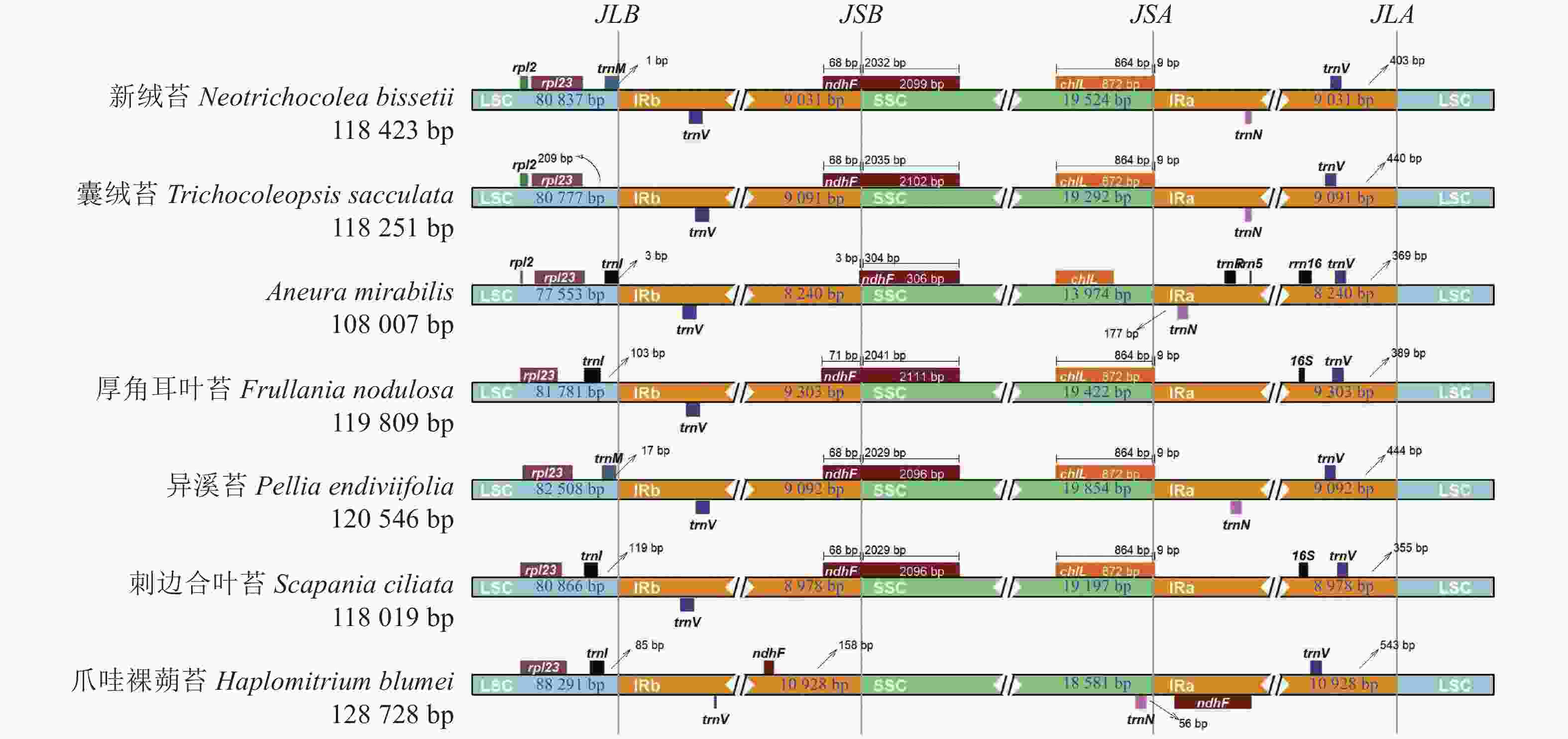
Figure 4. Chloroplast genome boundary analysis of 7 bryophytes
-
基于19种苔藓植物叶绿体基因组构建的BI树和ML树在拓扑结构上一致,各分支节点的支持率大部分为100%,可靠性较高。从系统发育树(图5)上看,叉苔亚纲的绿片苔和A. mirabilis最先分离出来,接着是溪苔亚纲的异溪苔,叶苔亚纲的14个种形成一支,内部包括2个姐妹分支:一支是广义叶苔目Jungermanniales s.l.,包括东亚鞭苔、护蒴苔、全萼苔、四齿异萼苔、中华羽苔和刺边合叶苔;另一支是广义光萼苔目Porellales s.l.和毛叶苔目Ptilidiales,前者包括白鳞苔、狭瓣疣鳞苔、厚角耳叶苔、毛耳苔、毛边光萼苔和日本扁萼苔,后者包括深裂毛叶苔、囊绒苔和新绒苔。新绒苔和囊绒苔形成1支,与深裂毛叶苔形成姐妹支,支持率为100%,说明其亲缘关系最密切。
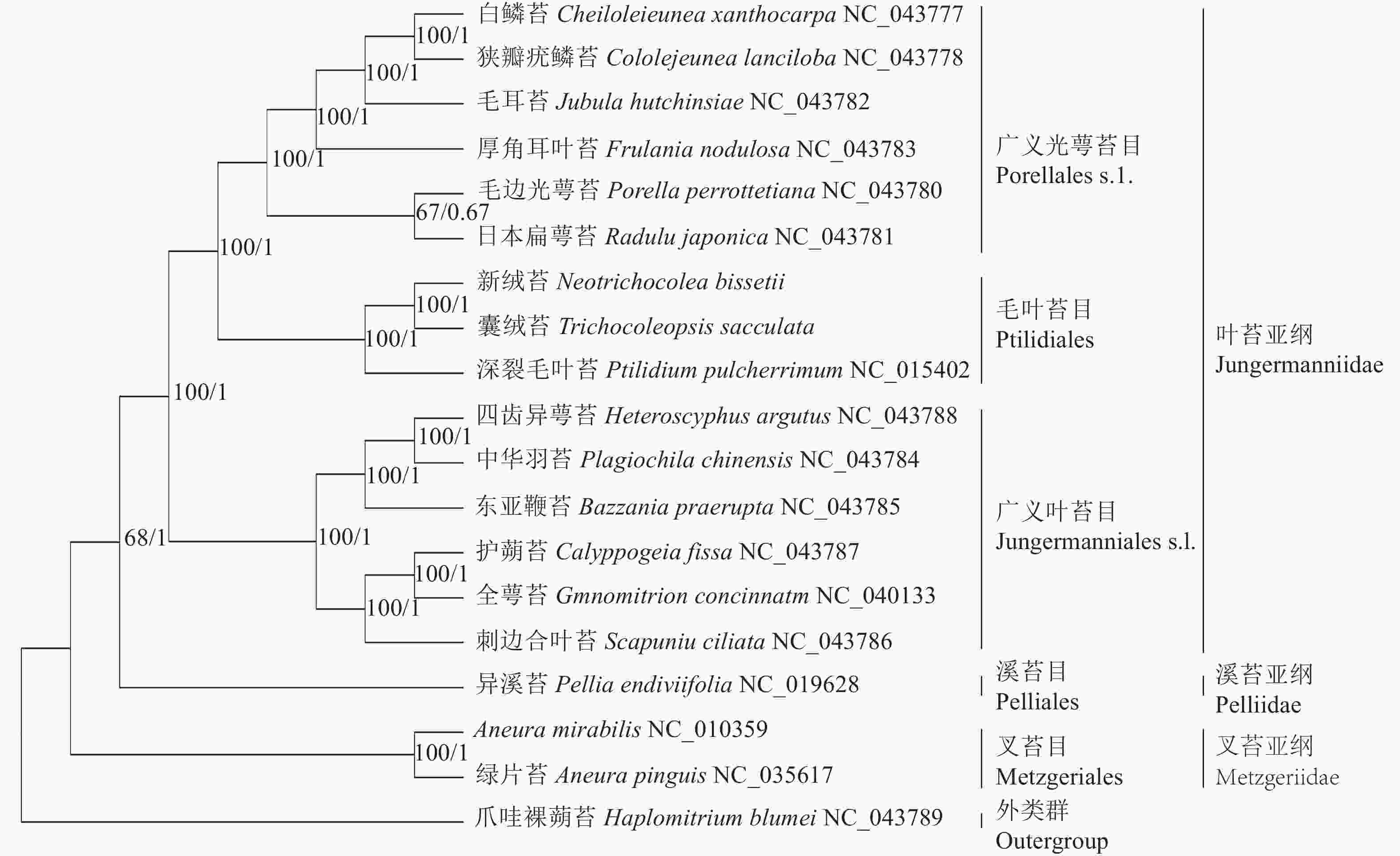
Figure 5. Phylogenetic tree based on chloroplast genome sequences of 19 bryophytes
-
新绒苔作为东亚特有的濒危苔藓植物,本研究首次对其叶绿体基因组进行测序、组装和分析。新绒苔叶绿体基因组总长度为118 423 bp,呈现典型的环状四分体结构,GC含量为33.3%,共编码了132个基因,与细光萼苔、巨瓣光萼苔、厚角耳叶苔、毛耳苔、尖瓣合叶苔Scapania ampliata植物叶绿体基因组特征相似[5, 29],表明苔藓植物叶绿体基因组进化相对保守。
SSR技术具有简便、快速、稳定性高和等位基因多样性高等特点,在基因组研究中,已经作为主要的分子标记技术广泛应用于比较基因组研究和系统学研究等领域[30]。重复序列分析显示:新绒苔叶绿体基因组有56个SSR,最丰富的类型是单核苷酸重复序列,单核苷酸重复序列以 A和T碱基为主。这与许多其他藓类植物物种报道的结果一致[7]。
植物在进化过程中,密码子的使用会受到自然选择、碱基突变、遗传漂变等因素的影响而产生偏好性[31]。植物叶绿体基因的RSCU可以确定密码子偏好性,RSCU≥1.00的密码子为高频率密码子,RSCU<1.00,则表示该密码子较少使用[32]。新绒苔 RSCU≥1.00的密码子有33个,其中以A、U结尾的密码子占84.8%,说明偏好以A、U结尾。与张家榕等[33]对18 种苔藓植物rbcL基因的密码子偏好分析结果相同。
IR和SSC边界的扩张和收缩导致相关基因拷贝数变化或边界区域假基因产生,这是叶绿体基因组进化过程中都存在的现象,也是其长度变化的主要原因[34]。本研究中,位于IR边界的基因包括trnV、trnL、ndhF、16S和chlL,且所有物种的IRb与SSC之间的连接位点均在ndhF基因内,且新绒苔的IR边界相对保守,只有极个别基因出现了缺失,如在JLB边界附近缺失trnL基因。新绒苔与囊绒苔同属于新绒苔科,叶绿体基因组特征和边界比较分析表明:除trnM基因外,其余基因位置一致,说明新绒苔与囊绒苔亲缘关系较近;二者在IRb均不含trnL基因,区别于其他目植物,说明二者与其余物种亲缘关系较远[35]。
本研究利用19种苔藓植物叶绿体基因组构建的系统发育树与近期研究一致[4-5, 36],支持了新绒苔科(包括新绒苔属和囊绒苔属)的成立,新绒苔科与毛叶苔科共同构成毛叶苔目,并与广义光萼苔目形成姐妹关系。
-
新绒苔的叶绿体基因组为典型的四分体结构,叶绿体全基因组序列全长为118 423 bp,包含79个蛋白质编码基因,8个rRNA和36个tRNA,系统发育分析显示新绒苔属和囊绒苔属为姐妹关系,共同构成了新绒苔科。本研究丰富了新绒苔属分子生物学资源,研究结果可为新绒苔及其近缘种的物种鉴定、资源保护和系统进化提供理论参考。
Characterization and phylogenetic location analysis of chloroplast of the endangered plant Neotrichocolea bissetii
doi: 10.11833/j.issn.2095-0756.20240356
- Received Date: 2024-05-16
- Accepted Date: 2024-09-14
- Rev Recd Date: 2024-09-09
- Available Online: 2025-01-20
- Publish Date: 2025-02-20
-
Key words:
- Neotrichocolea bissetii /
- chloroplast genome /
- characteristic analysis /
- phylogenetic
Abstract:
| Citation: | ZHU Mengfei, HU Yingfeng, SHI Xueqin. Characterization and phylogenetic location analysis of chloroplast of the endangered plant Neotrichocolea bissetii[J]. Journal of Zhejiang A&F University, 2025, 42(1): 55−63 doi: 10.11833/j.issn.2095-0756.20240356

|

 DownLoad:
DownLoad: