







-
果胶类多糖广泛分布于高等植物的根、茎、叶、果实等的细胞初生壁和细胞间隙,对细胞组织起着软化和黏合作用,同时还是抵御病原体微生物入侵的天然屏障[1]。2-酮-3-脱氧辛糖酸(KDO)是果胶类多糖鼠李半乳糖醛酸聚糖-Ⅱ(Rhamnogalacturonans Ⅱ,RG-Ⅱ)成分中很少见的八碳糖[2],在进化过程中非常保守,对花粉管的伸长和生长具有一定的作用[3-4]。KDO合成过程中涉及到5种酶[5],其中阿拉伯糖-5-磷酸异构酶(D-arabinose 5-phosphate isomerase,KdsD)是KDO生物合成过程中的第1个关键酶,可以催化核酮糖-5-磷酸(D-ribulose 5-phosphate,Ru5P)的异构化,以产生在KDO生物合成途径中的第1个中间产物D-阿拉伯糖-5-磷酸(D-arabinose 5-phosphate,A5P)。KDO最初是在革兰氏阴性菌中发现的,它连接O-特异侧链与类脂A共同嵌入细胞壁外膜上[6]。细胞壁中KDO合成的阻断会导致类脂A物质的累积和细胞生长停滞[7]。研究发现,KDO生物合成途径也存在于藻类植物与高等植物中[2, 8]。大肠埃希菌Escherichia coli中的KdsD晶体结构已经被解析,它是一个同源四聚体[9-11]。每个KdsD亚基包含2个不同的结构域:N-末端糖异构酶(SIS)结构域,主要参与磷糖异构化作用,以及1对未知功能的胱硫醚-β-合酶(CBS)结构域。通过定点突变确定了大肠埃希菌的Lys59,His88和His193对应于绿脓杆菌Pseudomonas aeruginosa的Lys56,His85和His190[12-14]分别是其活性重要的保守残基。目前,植物中KdsD的研究很少,其晶体结构也尚不清楚,植物来源的KdsD酶功能及催化作用机制、酶的催化特性等科学问题仍需要更加深入研究。毛竹Phyllostachys edulis是中国广泛分布的一种经济价值很高的植物,在林业生产中的地位至关重要。本研究拟克隆毛竹KdsD基因,分析它在不同组织的表达特异性,进行生物信息学分析和原核表达,经过Ni-NTA亲和层析和分子筛层析(SEC)纯化获得高纯度的蛋白,并对其酶学性质进行分析,为后续KdsD晶体学结构和植物KdsD基因功能的研究打下理论基础。
HTML
-
毛竹采自浙江农林大学亚热带森林培育国家重点实验室培育基地,取半年生毛竹实生苗新鲜的根、茎、叶分装于冻存管中,于液氮中速冻,放-80 ℃冰箱备用。
-
取毛竹新鲜的根、茎、叶在液氮中迅速研磨成粉末,用Trizol法[15]提取总RNA,用脱氧核糖核酸酶Ⅰ(DNaseⅠ)消除DNA污染。分别以毛竹约1.0 μg的根、茎、叶RNA为模板,利用逆转录试剂盒合成cDNA。
-
通过美国生物技术信息中心(NCBI)数据库获得拟南芥Arabidopsis thaliana的KdsD基因序列,检索毛竹数据库,得到一条同源序列。根据该基因的开放读码框(ORF)设计引物。上游BamH I酶切位点引物5′-CGGGATCCATGGGCTCGCTCCCCGTGCCCT-3′;下游Hind Ⅲ酶切位点引物5′-CCAAGCTTTTATAATCCAGCAGAGACCAAT-3′(酶切位点用下划线显示)。引物由上海生工生物工程股份有限公司合成。以反转录后的cDNA 为模板进行聚合酶链式反应(PCR)扩增。将PCR扩增产物电泳分析后经DNA胶回收试剂盒回收,获得的目的DNA片段插入经同样酶切后的pE-SUMO表达载体,得到1个N-末端含有6个His亲和标签的重组质粒。将重组质粒转化到大肠杆菌DH5α中,提取阳性克隆质粒进行双酶切和单酶切验证。
-
根据毛竹KdsD基因的保守序列设计qRT-PCR引物(正向引物:GTGTTGGATTCTGGATGGTTC;反向引物:CAGCCGTAGTGGTGAATGAGTAAC)。内参Actin(正向引物:CGACATTGGCTCGCTCTTC;反向引物:CGGCAGAGGTGAGGGAGAT),使用荧光定量PCR仪对PeKdsD基因在根、茎、叶不同组织的表达量进行分析。
-
将构建好的重组表达质粒转化到表达菌株E. coli(Ril)中,挑取单克隆,接入2 mL 溶菌肉汤(LB)培养基(A+)37 ℃培养,当菌体吸光度D(600)达到0.5~0.8时,加入终浓度为0.4 mmol·L-1的异丙基硫代半乳糖苷(IPTG)诱导,分别在37 ℃摇床上诱导3 h,在20 ℃摇床上过夜诱导,最后收集菌体,超声破碎,用十二烷基硫酸钠-聚丙烯酰氨凝胶电泳(SDS-PAGE)检测蛋白表达情况。
-
在确定的最佳温度表达条件下,将菌株接种于1.0 L LB培养基中进行扩大培养。加入诱导剂后,菌液在20 ℃,220 r·min-1条件下过夜培养。离心收集菌体,将裂解液[1.0 mol·L-1氯化钠,30.0 mmol·L-1三羟甲基氨基甲烷(Tris-HCl),体积分数为5%的甘油]加入到收集的菌体中,超声破碎细胞后,20 000 r·min-1,4 ℃条件下离心45 min,将上清转移至镍柱,置于摇床缓慢结合1 h,后分别用低浓度咪唑缓冲液洗脱杂蛋白,用高浓度咪唑缓冲液洗脱目的蛋白。将目的蛋白收集在10 kDa浓缩柱中离心进行浓缩,至体积为500.0 μL左右。使用分子筛层析柱SuperoseTM 12 10/300 GL上样,根据蛋白的出峰顺序收集蛋白,用SDS-PAGE胶检测蛋白的纯度,将较高纯度的蛋白收集浓缩,置于-80 ℃保存。
-
酶活力测定采用半胱氨酸-咔唑法[16-17]。25.0 μL的酶用100.0 mmol·L-1 Tris-HCl,pH 8.5配制,37 ℃保温3 min,加入25.0 μL 2.0 mmol·L-1 阿拉伯糖-5-磷酸(A5P),37 ℃反应5 min,加入50.0 μL 12.5 mol·L-1硫酸终止反应,采用半胱氨酸-咔唑法显色,测定D(540) nm吸光度。酶活力单位(1 U=16.67 nkat)定义为: 以A5P为底物,每分钟催化产生1.0 μmol的核酮糖-5-磷酸(Ru5P)所需的酶量。所有酶活测定结果均为3次重复平行试验数据的平均值。
-
最适pH值的测定,将样品KdsD稀释在100.0 mmol·L-1不同pH值缓冲液中,MES缓冲液从pH 6.0~6.5,Tris-HCl缓冲液从pH 7.0~9.0。37 ℃反应5 min,测定酶活。最适温度则是在测定的最适pH值下,将反应温度控制在25~65 ℃,隔10 ℃进行酶促反应,然后测定酶活性。
1.1. 实验材料
1.2. 实验方法
1.2.1. 总RNA提取和cDNA合成
1.2.2. PeKdsD基因克隆
1.2.3. 定量反转录聚合酶链式反应(qRT-PCR)分析
1.2.4. PeKdsD的原核表达
1.2.5. PeKdsD的纯化
1.2.6. 酶活性测定
1.2.7. 最适pH值和最适温度对酶活性的影响[17]
-
PeKdsD基因经PCR扩增重组后测序,得到1条开放读码框(ORF)为1 038 bp的目的片段。该基因编码346个氨基酸(图 1)。将PCR产物与pE-SUMO表达载体连接,产物转化到DH5α,挑取阳性菌落进行菌检测出,成功构建原核表达载体。

Figure 1. ORF of PeKdsD gene and the corresponding amino acid sequence
-
通过blast在线软件分析,结果显示,PeKdsD含有2个不同的结构域:SIS结构域和CBS结构域,大肠埃希菌序列中关键的催化残基Lys59,His88和His193在毛竹中是完全保守的。PeKdsD编码的氨基酸序列与玉米Zea mays,高粱Sorghum bicolor和水稻Oryza sativa具有较高的一致性,均为91.00%,与杨树Populus trichocarpa和拟南芥的一致性分别为69.00%和66.38%,而与大肠埃希菌和脆弱拟杆菌Bacteroides fragilis的一致性分别只有29.21%和31.12%(图 2)。采用MEGA6软件构建不同物种KdsD氨基酸序列的系统进化树(图 3),结果表明:毛竹与玉米位置较近,位于同一个分支上;与杨树、水稻位于同一大分支上,与拟南芥和脆弱拟杆菌分支较远。

Figure 2. Sequence multiple alignment of KdsD from different species

Figure 3. Molecular phylogenetic tree of KdsD from different species
-
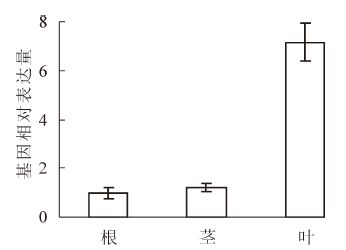
PeKdsD不同组织qRT-PCR结果显示:PeKdsD在毛竹的根、茎、叶中均有表达,但在表达量上存在较大的差异,在叶中的表达量远远大于根与茎(图 4)。

Figure 4. Relative expression of PeKdsD in the root, shoot and leaf of Phyllostachys edulis
-
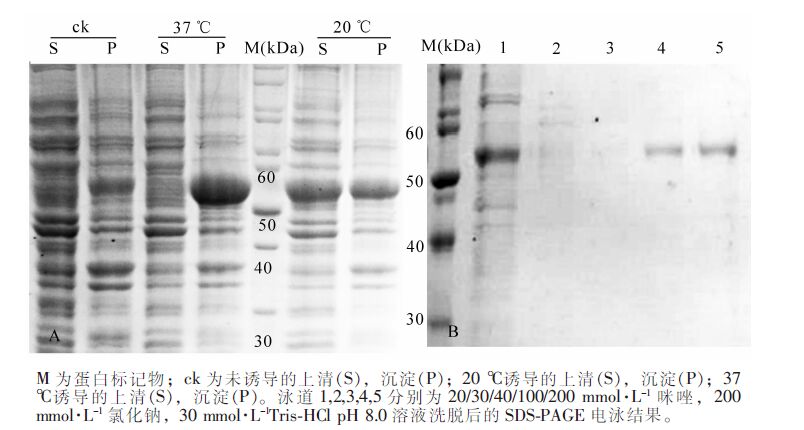
将PeKdsD重组载体转化到表达菌株E. coli(Ril)感受态细胞,使用IPTG诱导表达,SDS-PAGE检测蛋白表达及可溶性情况(图 5A)。对比发现表达菌株Ril在20 ℃培养温度下表达量相对较高,故在后续试验中确定重组蛋白诱导表达的最佳温度为20 ℃。

Figure 5. Prokaryotic expression of PeKdsD in Escherichia coli(Ril) cell (A) and SDS-PAGE analysis from Ni-NTA affinity chromatography of PeKdsD (B)
-
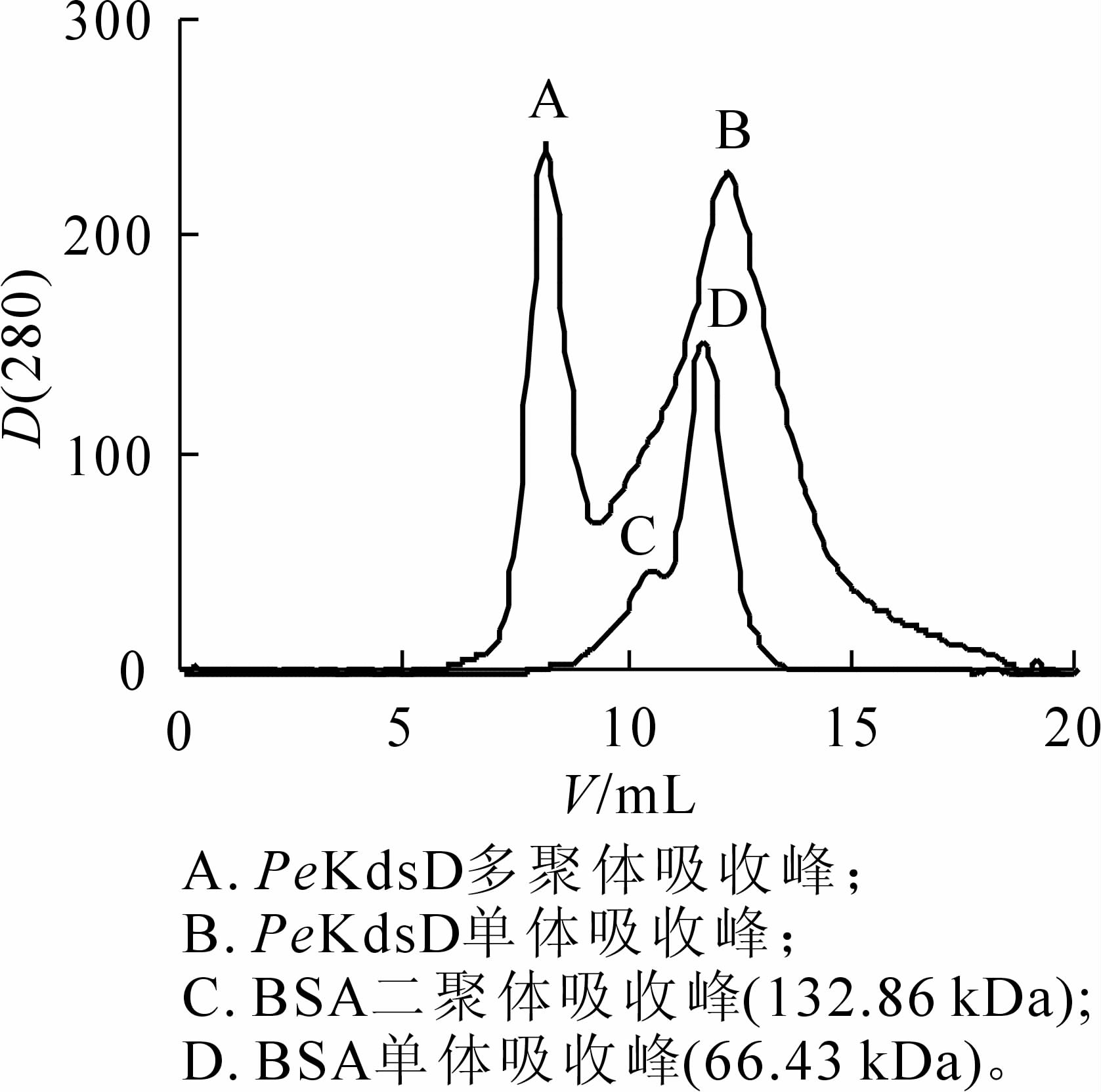
将收集的菌体超声破碎离心后,上清加入Ni-NTA柱,冰上结合后,采用咪唑梯度洗脱蛋白,SDS-PAGE检测(图 5B)。结果显示:条带出现的位置与目的蛋白大小一致,目的蛋白被成功地纯化出来。将目的蛋白进一步经SuperoseTM 12 10/300 GL分子筛层析后,出现2个明显的吸收峰。对比小牛血清BSA出峰位置(二聚体C峰对应分子量为132.86 kDa,单聚体D峰对应分子为66.43 kDa),A峰对应是KdsD多聚体,B峰对应的是KdsD单体(图 6),说明KdsD蛋白在溶液中存在多种聚体形式。2步纯化Ni-NTA结合SEC可分离纯化得到不同状态的高纯度KdsD蛋白,可满足今后酶学特性的研究和蛋白晶体生长的要求。

Figure 6. Purification of PeKdsD by size exclusion chomatography
-
酶活性测定结果表明:纯化的KdsD酶最适温度为37 ℃,在35~45 ℃范围内酶活力较高,45 ℃以上酶活力急剧下降,而在25 ℃时仍保持在60%以上(图 7A);不同的pH值条件也会影响酶的活性,最适作用pH值为pH 8.5,此时活性较高,过酸或过碱环境都会对酶活性有较大的抑制作用(图 7B)。

Figure 7. Effect of temperature and pH value on activity of enzyme
2.1. PeKdsD基因克隆及原核表达载体的构建
2.2. PeKdsD氨基酸序列比较与系统进化树分析


2.3. PeKdsD在不同组织中的表达分析
2.4. PeKdsD重组蛋白的纯化及表达检测
2.5. PeKdsD蛋白Ni-NTA纯化与分子筛层析
2.6. 重组酶的酶学性质

-
自2010年大肠埃希菌的KdsD结构公布后,又陆续有其他微生物的KdsD结构报道。2014年CHIU等[14]解析出了脆弱拟杆菌API(bfAPI)的晶体结构,为四聚体。但是植物来源的KdsD相关研究甚少,其结构和功能至今仍未被解析。本研究首次成功克隆到了毛竹KdsD基因,该基因具有一个完整的开放阅读框(ORF),编码346个氨基酸。生物信息学分析预测其含有2个不同的结构域:N-末端糖异构酶(SIS)结构域和胱硫醚-β-合酶(CBS)结构域。尽管大肠埃希菌序列中关键的催化残基Lys59,His88和His193在毛竹中是完全保守的,但是毛竹KdsD蛋白序列与大肠埃希菌KdsD蛋白序列相似度不足30%,因此,推测植物KdsD与微生物中的KdsD蛋白空间结构和功能可能存在一定的差异。为进一步了解植物KdsD和微生物KdsD结构上的差异,还需进一步纯化获得高纯度的蛋白,进行蛋白结晶,通过X射线等方面的研究进一步分析验证结构与功能的关系。实时定量PCR在根、茎、叶中均检测到有表达,尤其在叶中表达量比其他组织高约5~8倍,表现出明显的组织特异性现象。Ni-NTA纯化显示所得的目的蛋白大小与预测的蛋白分子量相一致,成功纯化出了PeKdsD蛋白。进一步的SEC纯化结果表明:毛竹KdsD在30 mmol·L-1 Tris-HCI pH 8.0,200 mmol·L-1氯化钠溶液条件,以多聚体混合的形式存在,SEC中KdsD的多聚体究竟是几聚体还需分析超速离心(AUC)等实验做进一步的分析验证。酶学性质初步测定结果表明:PeKdsD的最适温度为37 ℃,最适pH值为pH 8.5。最适pH值与预测的pH值有所偏差,故后期试验采用pH 8.5为宜。当然,要揭示毛竹KdsD酶功能及催化作用机制,还有待于后续晶体结构与功能等方面的进一步深入研究。
 DownLoad:
DownLoad: