






-
重瓣榆叶梅Prunus triloba ‘Multiplex’属蔷薇科Rosaceae李属Prunus灌木或小乔木,早春开花,是中国特有园林植物[1−2]。重瓣榆叶梅具有较强的抗盐碱能力,广泛栽培于中国东北、华北和西北地区[3];重瓣榆叶梅花和花粉富含蛋白质、生物酶、多糖、脂肪、氨基酸和维生素等物质,可以用作保健品、药品、食品和营养性化妆品的原材料[4−5]。种仁含油量较高,可代郁李仁入药[6]。重瓣榆叶梅于1855年被引入欧洲、美洲、大洋洲[7],广泛用于园林绿化。研究发现:榆叶梅P. triloba与近缘种桃P. persica的核不对称性较接近,榆叶梅为八倍体(2n=64),核型分类为2B或2A,核不对称系数高达63.08%[2]。但榆叶梅的物种发生还尚不清楚,关于重瓣榆叶梅和榆叶梅系统发育关系仍是空白。
叶绿体基因组是植物叶绿体的独立遗传系统,重组率低且保守,一般为四分体结构,由大单拷贝区(LSC)、双向重复区(IR)和小单拷贝区(SSC)组成,在植物物种鉴定、物种分化、DNA条形码开发和系统分类研究中具备可信度[8-11]。已有研究通过对46个类群的叶绿体atpB-rbcL构建进化树,明确了小麦族猬草属Hystrix和赖草属Leymus的系统发育关系[12];基于rbcL + matK + trnH-psbA组合序列和叶绿体全基因组研究白头翁属Pulsatilla物种的系统发育关系,发现变异热点在物种识别和系统发育分析中建树结果相似[13];基于叶绿体psbD 基因构建的进化树表明:花叶矢竹Pseudosasa japonica f. akebonosuji与毛竹Phyllostachys edulis的亲缘关系较近[14]。目前,已获得野生榆叶梅叶绿体基因组序列[3],但未见关于重瓣榆叶梅的叶绿体基因组序列报道。本研究通过 Illumina NovaSeq高通量测序平台,成功组装了重瓣榆叶梅叶绿体全基因组序列结构并分析其遗传多样性,以期为重瓣榆叶梅系统发育、物种鉴定、资源开发研究提供理论依据。
-
2020和2021年3—4月,对河南省洛阳市洛龙区(34°38′09.94″N,112°26′36.61″E)重瓣榆叶梅种质资源进行调查,采集重瓣榆叶梅植株的幼嫩新鲜叶片,用纯净水和纸巾依次清洁叶片表面,装入有变色硅胶颗粒的密封袋,带回实验室保存以备用。
-
采用 2×CTAB法提取重瓣榆叶梅总DNA[15]。叶片于液氮中研磨后用CTAB法提取植物总DNA。以琼脂糖凝胶电泳(琼脂质量分数为1%,电压为120 V,电泳时间为25 min)和微量核酸测定仪(NanoDrop 2000)检测样本DNA的完整性、质量和质量分数。样本DNA质量浓度为56.16 mg·L−1,体积为40 μL,总量为2.25 μg,D(260/280)为1.89,D(260/230)为1.02,检测结果为A类。样品质量满足建库测序要求,且总量满足2次或2次以上建库需要。
-
已检测为A类的DNA样品用超声波机械打断,经片段纯化、末端修复、3′加 A、连接测序接头后,进行PCR 扩增构建文库,在Illumina NovaSeq平台进行测序得到原始序列(raw reads),对原始序列进行过滤:去除带接头的序列,过滤N含量超过10%的序列,通过BLAST 软件比对核酸序列数据库(NT)等质控得到过滤序列(clean reads)。原始序列上传于数据库GeneBank。
使用软件SPAdes[16]、aragorn v1.2.38、DOGMA[17]和CPGview-RSG[18]进行测序数据组装和预测注释重瓣榆叶梅叶绿体的基因,通过手工纠正,成功获得其全叶绿体基因组序列并完成重瓣榆叶梅叶绿体基因组的基因注释。用在线软件(
https://chlorobox.mpimp-golm.mpg.de/OGDraw.html )绘制其叶绿体全基因组物理可视化图谱。 -
用CodonW 1.4.2对重瓣榆叶梅叶绿体基因组密码子数量和相对同义密码子使用度(relative synonymous codon usage,RSCU)进行分析。
-
利用MISA-web[19]检测重瓣榆叶梅的cpSSR位点,参数设置为单核苷酸-8、双核苷酸-5、三核苷酸-3、四核苷酸-3、五核苷酸-3和其他核苷酸-3。
-
于美国国家生物技术信息中心(NCBI)数据库中下载MK764428、HQ336405、MK798146、KF990036、KY101152、MK790138、MG602257、MK434918和MN661138等多个物种的叶绿体基因组序列,构建包括重瓣榆叶梅在内的蔷薇科李属植物的系统发育进化树,观察并分析重瓣榆叶梅和其他物种间的亲缘关系和系统发育地位。系统发育进化树采用全叶绿体基因组数据进行分析:用MAFFT[20](v7.427,auto模式)进行多序列比对,将比对好的数据用trimAl v1.4.rev15修剪,使用RAxML v8.2.10,选用GTRGAMMA模型,bootstrap=1 000,构建最大似然进化树。
-
重瓣榆叶梅全叶绿体基因组总长度为157 827 bp,数据已上传至 GeneBank,登录号为MT937181。基于Illumina NovaSeq 6000测序平台表明,重瓣榆叶梅(MT937181)的读取总和(ReadSum)、基本总和(BaseSum)分别为15 307 141、4 592 142 300 bp,Q20和Q30分别为97.91%和93.90%。重瓣榆叶梅全叶绿体基因组为经典的四分体结构,由1个大单拷贝区域(LSC),1个小单拷贝区域(SSC)及反向重复区域(IRa/IRb)构成(表1~2和图1),其序列长度分别为86 032、19 023、26 386 bp。重瓣榆叶梅全叶绿体基因组中GC和AT的总占比分别为 36.80%和63.20%,GC占比在 IR、LSC 和SSC区域存在较大的差异,IR区域GC占比最高可达42.58%;LSC 次之,GC占比34.64%;SSC中GC占比最低仅30.48%(表1)。
区域 全叶绿体基因组 大单拷贝区(LSC) 小单拷贝区(SSC) 反向重复区(IRa) 反向重复区(IRb) 数量 占比/% 数量 占比/% 数量 占比/% 数量 占比/% 数量 占比/% A 49 091 31.10 27 334 31.77 6 606 34.73 7 588 28.76 7 563 28.66 C 29 663 18.79 15 381 17.88 3 047 16.02 5 416 20.53 5 819 22.05 G 28 410 18.00 14 423 16.76 2 752 14.47 5 819 22.05 5 416 20.53 T 50 663 32.10 28 894 33.59 6 618 34.79 7 563 28.66 7 588 28.76 GC 58 073 36.80 29 804 34.64 5 799 30.48 11 235 42.58 11 235 42.58 总计 157 827 100.00 86 032 100.00 19 023 100.00 26 386 100.00 26 386 100.00 Table 1. Structure and composition of P. triloba ‘Multiplex’ chloroplast genome
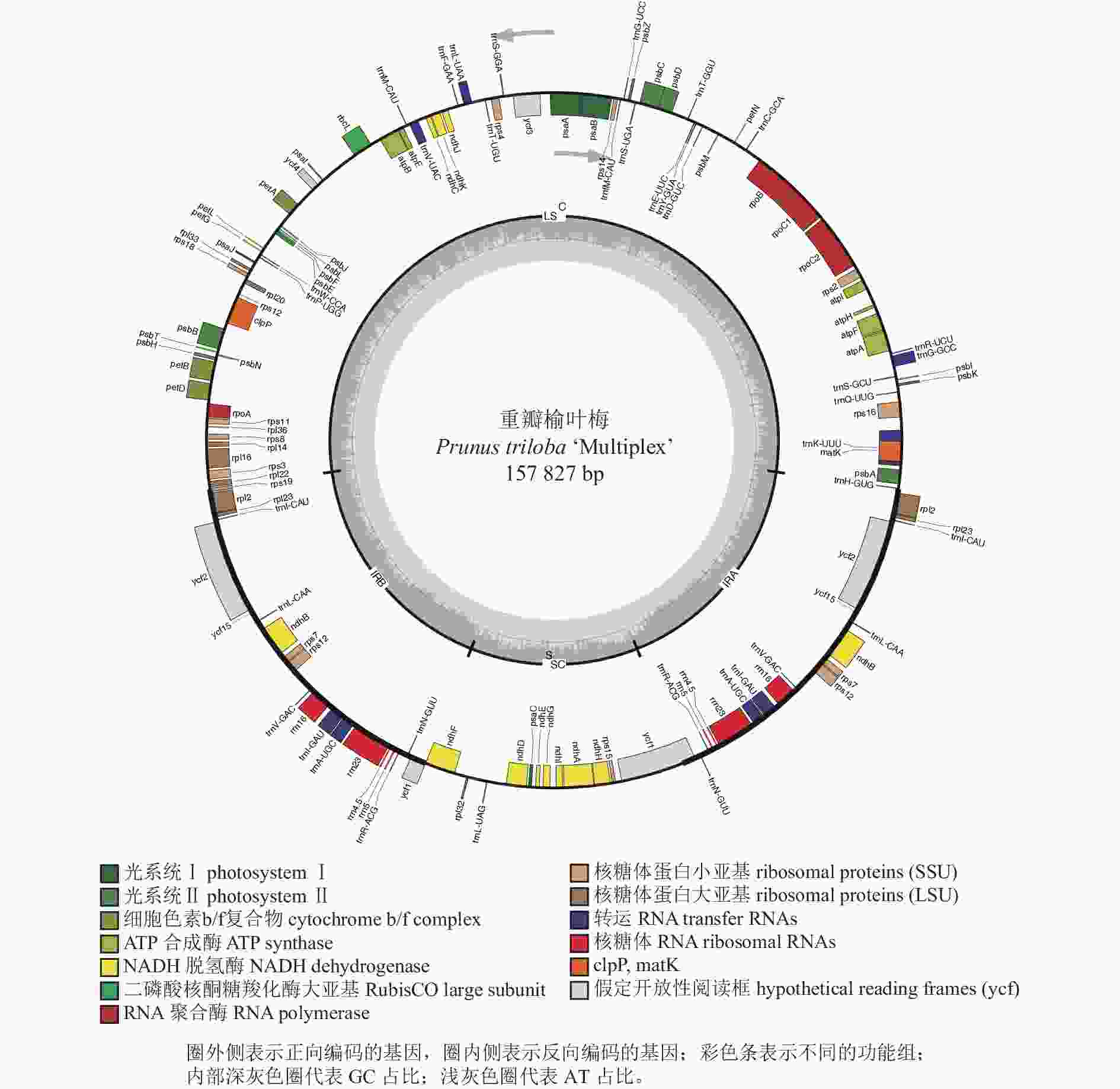
Figure 1. Gene map in P. triloba ‘Multiplex’ chloroplast genome
重瓣榆叶梅全叶绿体基因组中注释了132个基因(图1和表2),LSC区、IR区和SSC区的基因数量分别为82个(62.12%)、19个(14.39%)和12个(9.09%),蛋白编码基因有87个,占比65.91%,核糖体RNA(rRNA)数量为8个,占比6.06%,转运RNA(tRNA)数量为37个,占比28.03%。重瓣榆叶梅全叶绿体基因组序列的基因中, ycf2长度为6 834 bp,ycf1为5 589 bp,rpoC2为4 107 bp,是排名在前3位的最大基因。rRNA 基因rrn4.5、rrn16、rrn5、rrn23在IRs区,均有2个拷贝,其基因长度分别为103、1 491、121和2 809 bp。
重瓣榆叶梅全叶绿体基因组与其他植物的叶绿体基因类似,大多数基因均不存在内含子且基因数量为1。重瓣榆叶梅全叶绿体基因组有19个双拷贝类型基因,占比为14.39%,包括:5个自我复制类型基因,即ndhB(NADH脱氢酶亚基基因)、rpl2、rpl23、rps12、rps7;4个rRNA基因,即rrn16、rrn23、rrn4.5、rrn5;7个tRNA基因,即trnA-UGC、trnI-CAU、trnI-GAU、trnL-CAA、trnN-GUU、trnR-ACG、trnV-GAC;3个未知功能蛋白基因,即ycf1、ycf2和ycf15。除atpF、ndhA、ndhB、petB、petD、rpl16、rpl2、rpoC1、rps12、rps16、trnA-UGC、trnG-GCC、trnI-GAU、trnK-UUU、trnL-UAA、trnV-UAC等16个基因各含1个内含子,clpP、ycf3基因含2个内含子,其余大部分基因不含内含子(表2)。
基因分类 基因分组 基因名称 光合作用相关基因 光合系统Ⅰ基因 psaA、psaB、psaC、psaI、psaJ 光合系统Ⅱ基因 psbA、psbB、psbC、psbD、psbE、psbF、psbH、psbI、psbJ、psbK、psbL、psbM、psbN、psbT、psbZ NADH脱氢酶基因 ndhA*、ndhB*(2)、ndhC、ndhD、ndhE、ndhF、ndhG、ndhH、ndhI、ndhJ、ndhK 细胞色素复合物基因 petA、petB*、petD*、petG、petL、petN ATP合酶基因 atpA、atpB、atpE、atpF*、atpH、atpI 二磷酸核酮糖羧化酶大亚基基因 rbcL 自我复制相关基因 核糖体大亚基基因 rpl14、rpl16*、rpl2*(2)、rpl20、rpl22、rpl23(2)、rpl32、rpl33、rpl36 核糖体小亚基基因 rps11、rps12**(2)、rps14、rps15、rps16*、rps18、rps19、rps2、rps3、rps4、rps7(2)、rps8 RNA聚合酶亚基基因 rpoA、rpoB、rpoC1*、rpoC2 核糖体RNA基因 rrn16(2)、rrn23(2)、rrn4.5(2)、rrn5(2) 转运RNA基因 trnA-UGC*(2)、trnC-GCA、trnD-GUC、trnE-UUC、trnF-GAA、trnG-GCC*、trnG-UCC、trnH-GUG、trnI-CAU(2)、trnI-GAU*(2)、trnK-UUU*、trnL-CAA(2)、trnL-UAA*、trnL-UAG、trnM-CAU、trnN-GUU(2)、trnP-UGG、trnQ-UUG、trnR-ACG(2)、trnR-UCU、trnS-GCU、trnS-GGA、trnS-UGA、trnT-GGU、trnT-UGU、trnV-GAC(2)、trnV-UAC*、trnW-CCA、trnY-GUA、trnfM-CAU 其他基因 成熟酶基因 matK 依赖ATP的蛋白酶单元p基因 clpP** 包裹膜蛋白基因 cemA 乙酰辅酶A羧化酶亚基基因 accD c型细胞色素合成基因 ccsA 未知功能基因 保守开放阅读框 ycf1(2)、ycf15(2)、ycf2(2)、ycf3**、ycf4 说明:* 代表基因有1个内含子;**代表基因有2个内含子;(2)代表基因的拷贝数为2。 Table 2. List of genes present in P. triloba ‘Multiplex’ chloroplast genome
-
MT937181中发现共有26 678个编码密码子(表3)。其中,亮氨酸(Leu)的编码密码子数量最多,达2 792个;蛋氨酸(Met)的编码密码子有AUG、AUU、CUG、GUG和UUG等,其中AUU和UUG数量最少,均只有1个。重瓣榆叶梅全叶绿体基因组的编码异亮氨酸(Ile)的AUU数量最多,达1 108个。编码Leu的密码子最多,占总量的10.47%;而最少的色氨酸(Trp)仅占1.70%。此外,31种密码子的相对同义密码子使用度(RSCU)>1,表明它们在重瓣榆叶梅全叶绿体基因组中是偏好密码子。在偏好密码子中有29种都是以A或者U结尾,占RSCU>1的密码子总量93.55%,只有编码Leu的UUG和编码Met的AUG这2种偏好密码子是以G结尾,偏好密码子多选择密码子第3位是 A或者T,体现出密码子A/T偏好性。RSCU<1的密码子有36个,其中以G/C碱基结尾的有33个,说明RSCU<1的重瓣榆叶梅全叶绿体基因组的密码子更倾向以G/C碱基结尾;Trp的RSCU=1,无密码子偏好性。
符号 氨基酸 密码子 数量/个 RSCU 符号 氨基酸 密码子 数量/个 RSCU * Ter UAA 52 1.793 1 M Met GUG 2 0.016 0 * Ter UAG 21 0.724 2 M Met UUG 1 0.008 0 * Ter UGA 14 0.482 7 M Met AUG 620 0.495 2 A Ala GCA 385 1.096 8 N Asn AAC 308 0.470 2 A Ala GCC 226 0.644 0 N Asn AAU 1002 1.529 8 A Ala GCG 155 0.441 6 P Pro CCA 313 1.137 2 A Ala GCU 638 1.817 6 P Pro CCC 208 0.755 6 C Cys UGC 78 0.500 0 P Pro CCG 151 0.548 4 C Cys UGU 234 1.500 0 P Pro CCU 429 1.558 4 D Asp GAC 213 0.393 4 Q Gln CAA 725 1.547 4 D Asp GAU 870 1.606 6 Q Gln CAG 212 0.452 6 E Glu GAA 1 041 1.476 6 R Arg AGA 503 1.880 4 E Glu GAG 369 0.523 4 R Arg AGG 176 0.658 2 F Phe UUC 524 0.688 2 R Arg CGA 358 1.338 6 F Phe UUU 999 1.311 8 R Arg CGC 111 0.415 2 G Gly GGA 728 1.621 2 R Arg CGG 118 0.441 0 G Gly GGC 174 0.387 6 R Arg CGU 339 1.267 2 G Gly GGG 303 0.674 8 S Ser AGC 140 0.412 8 G Gly GGU 591 1.316 4 S Ser AGU 398 1.174 2 H His CAC 152 0.471 4 S Ser UCA 414 1.221 0 H His CAU 493 1.528 6 S Ser UCC 327 0.964 8 I Ile AUA 732 0.959 1 S Ser UCG 187 0.551 4 I Ile AUC 450 0.589 5 S Ser UCU 568 1.675 8 I Ile AUU 1 108 1.451 4 T Thr ACA 422 1.241 2 K Lys AAA 1 080 1.505 2 T Thr ACC 250 0.735 2 K Lys AAG 355 0.494 8 T Thr ACG 151 0.444 0 L Leu CUA 367 0.788 4 T Thr ACU 537 1.5796 L Leu CUC 186 0.399 6 V Val GUA 557 1.537 6 L Leu CUG 188 0.403 8 V Val GUC 161 0.444 4 L Leu CUU 580 1.246 2 V Val GUG 206 0.568 8 L Leu UUA 909 1.953 6 V Val GUU 525 1.449 2 L Leu UUG 562 1.207 8 W Trp UGG 454 1.000 0 M Met AUU 1 0.008 0 Y Tyr UAC 205 0.400 0 M Met CUG 2 0.016 0 Y Tyr UAU 820 1.600 0 Table 3. Codon usage of P. triloba ‘Multiplex’
-
采用MISA软件对重瓣榆叶梅叶绿体基因组进行了SSR分析(图2和表4)。在重瓣榆叶梅叶绿体基因组中共查找到 236个符合条件的SSR位点。单核苷酸重复单元有146个,双核苷酸重复单元有12 个,三核苷酸重复单元有62个,四核苷酸重复单元有8个,五核苷酸重复单元有1个,其他核苷酸重复单元有7个。叶绿体不同区域的SSR分布情况有差异,大部分SSR位于LSC,一部分在 IRs 区域内,少数位于 SSC区域。分布在IR、LSC、SSC区域的SSR位点分别为38、158、40个,占比分别为16.10%、66.95%、16.95%。SSR位点在不同功能的基因间分布也不均:114个位于基因间隔区(IGS), 82个SSR位于外显子,40个SSR位于内含子。从分布情况发现,SSR多位于 LSC和基因间隔区。从碱基的组成上看,SSR 中重 复 占 比最 大 的 是 单核苷 酸 , 约 61.86%。重瓣榆叶梅叶绿体基因组中的SSR主要是由A和 T组成的,占所有重复序列的 72.46%,其中 A/T 碱基构成的单核苷酸重复序列137条,AT/TA组成的二核苷酸重复序列 11条,A与T组成的三核苷酸重复序列18 条,A与T 组成的四核苷酸重复序列5条。
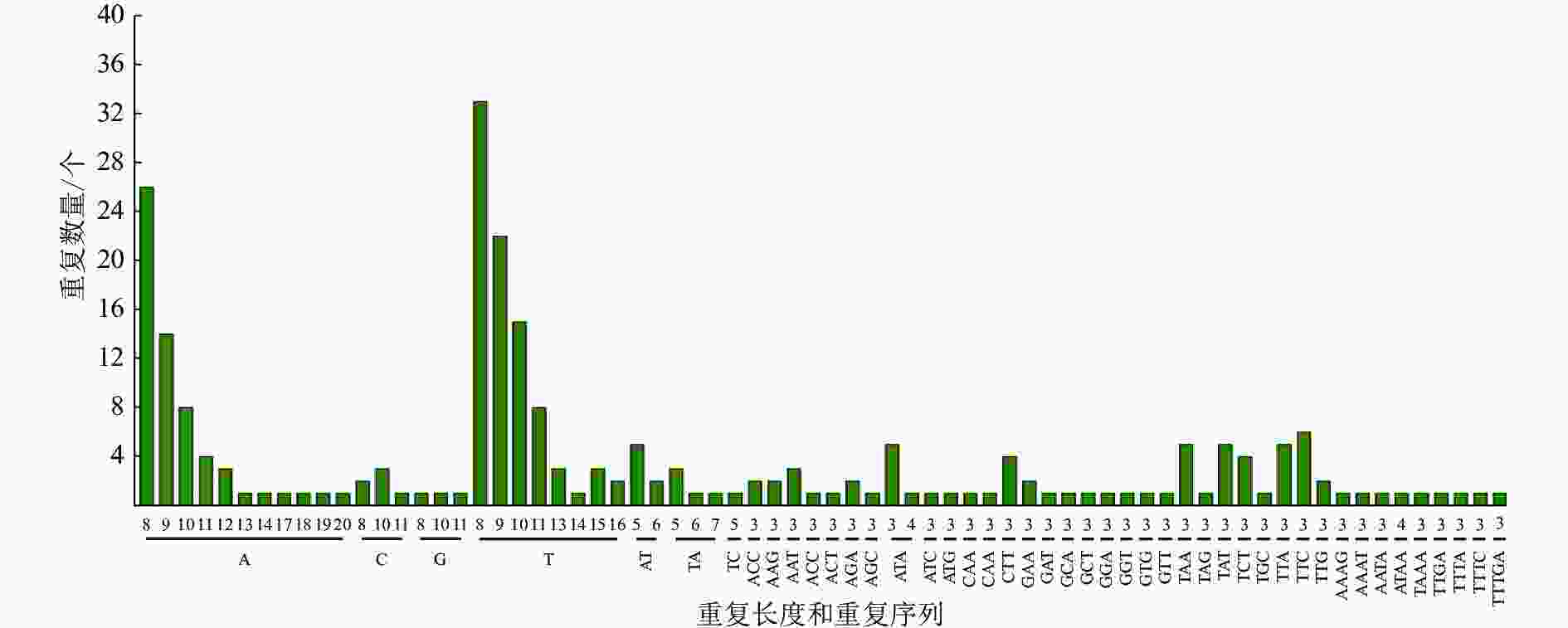
Figure 2. Type and number of SSR loci of P. triloba ‘Multiplex’ chloroplast genome
区域 数量/个 占比/% 外显子/个 内含子/个 基因间区/个 IR 38 16.10 21 4 13 LSC 158 66.90 35 33 90 SSC 40 16.90 26 3 11 总计 236 100.00 82 40 114 Table 4. Region of SSR loci of P. triloba ‘Multiplex’ chloroplast genome
重瓣榆叶梅SSR位点在叶绿体基因组中多态性较高,具有A/T碱基偏好性。SSR主要是A或T、ATA、ATTA/TAAT/AT复合重复、TTA /TAT或AT/TA,为SSR分子标记的开发打下基础。
-
选择重瓣榆叶梅与东京樱花P. yedoensis、扁桃P. dulcis、麦李P. glandulosa 和中国李P. salicina的叶绿体基因组进行Mauve比对(图3),结果表明:重瓣榆叶梅全叶绿体基因组与其余4个物种序列有良好的共线性关系,叶绿体基因组未检测到大片段的基因重排。5个物种的基因组序列横向均分为3个部分(各物种序列长度与图中序列长度不同),从左到右为LSC/IR/SSC,纵向有一直竖线,说明5个物种叶绿体基因组的基因组成和排列十分相似。LSC和SSC中粉色局块斑纹较多,白色多为IR区,几乎为空白,说明IR区域十分保守。76 915~81 087 bp局块区间,5个物种的对比差异性最大。

Figure 3. Genomic collinearity of P. triloba ‘Multiplex’ chloroplast genome
重瓣榆叶梅全叶绿体基因组与已经公布的其他植物的叶绿体序列构建系统进化树(图4)。结果发现:进化树上各节点和分支聚类的支持率较高,检验分值可达 100% ,说明聚类结果的可靠性较高。由聚类图可知重瓣榆叶梅与榆叶梅聚合在一个分支,和长柄扁桃P. pedunculata亲缘关系最近。基于单拷贝核基因 RPB2、Leafy内含子序列对榆叶梅及其近缘种系统发育学分析,发现桃P. persica、普通扁桃P. dulcis、蒙古扁桃P. mongolica 聚为一个类群;榆叶梅、长柄扁桃、矮扁桃P. tenella聚为一个类群。矮扁桃的系统发育地位有争议,可能与矮扁桃的取样和命名有极大关系,需要进一步探讨。核基因和叶绿体基因组共同认为榆叶梅和长柄扁桃具有同源最近亲缘关系,在进行榆叶梅品种选育和育苗育种时,可充分考虑榆叶梅与长柄扁桃的系统发育关系。

Figure 4. Phylogenetic tree of P. triloba‘Multiplex’ and its related species
-
被子植物的叶绿体基因组长度约1.60×105 bp,编码基因数约130个[21−22]。本研究获得的重瓣榆叶梅全叶绿体基因组序列MT937181全长为 157 827 bp,编码基因数为132个,与一般被子植物相符合。重瓣榆叶梅为园林栽培植物,分布广泛;其他种为野生种,分布和自然生境不同,其中蒙古扁桃十分耐寒且仅分布于蒙古高原。将4个不同叶绿体基因组序列进行比较,发现重瓣榆叶梅总序列、LSC和SSC长度最小,蒙古扁桃SSC和IR长度最大。推测可能在人工栽培和植物品种选育中,重瓣榆叶梅由于外在表现性状和生长环境的优越,其叶绿体基因组中基因种类增加,但总序列长度减小;而在野生环境分布狭窄或需要极端耐寒时,蒙古扁桃为适应不良环境,其SSC区和IR长度增大,基因种类和数量发生较大变化,ycf15基因缺失,且增加了1个rps19基因。ycf15属于未知功能基因,在龙葵属Amborella和蔷薇类等原始被子植物中无功能,在毛茛属Ranunculus植物已经完全丢失[23]。rps4以及rps19为核糖体小亚基类基因,核糖体在细胞中负责完成“中心法则”里“翻译”:由RNA到蛋白质这一过程。核糖体小亚基通过与信使RNA结合,转运RNA运送的氨基酸分子来合成多肽。ycf15、rps4以及rps19等3类基因的变化情况可能与植物对栽培选育、不同环境适应有直接关系。
自然界遗传密码子在mRNA翻译成蛋白质时具有重要决定作用。同义密码子指编码同一种氨基酸的不同密码子,有时具有使用偏好性[10, 24]。重瓣榆叶梅编码基因RSCU>1密码子的第 3个核苷酸具有明显的A/U偏好。简单重复序列(SSR)广泛分布于真核生物基因组中,重瓣榆叶梅与榆叶梅(MK790138)、长柄扁桃(MG602257)和蒙古扁桃(MG602256)叶绿体基因组的SSR位点均具有富集多聚A或多聚T情况。本研究系统进化分析表明:重瓣榆叶梅与榆叶梅、长柄扁桃亲缘关系最近,进化结果与传统分类学相符[2−3, 24−26]。
-
本研究成功获得了重瓣榆叶梅全叶绿体基因组序列,其总长度为157 827 bp,具备经典的四分体结构,GeneBank登录号为MT937181,编码基因数为132个。LSC、SSC和IRa/IRb序列长度分别为86 032、19 023和26 386 bp,GC占比在 IR、LSC 和SSC区域存在较大的差异。重瓣榆叶梅叶绿体基因组与其他3种近缘种植物比较中,ycf15、rps4以及rps19等3类基因有较大变化,可能是栽培环境的选择影响。重瓣榆叶梅叶绿体基因组的SSR位点具有富集A/T情况。基于叶绿体基因组数据确定了重瓣榆叶梅和榆叶梅系统发育关系,系统进化分析结果与传统植物分类学与形态学相符,即重瓣榆叶梅与榆叶梅、长柄扁桃亲缘关系最近。
Genetic characteristics of whole chloroplast genome in Prunus triloba ‘Multiplex’
doi: 10.11833/j.issn.2095-0756.20230489
- Received Date: 2023-09-30
- Accepted Date: 2024-03-06
- Rev Recd Date: 2023-12-13
- Available Online: 2024-05-22
- Publish Date: 2024-05-22
-
Key words:
- Prunus triloba ‘Multiplex’ /
- whole chloroplast genome /
- codon preference /
- repeated sequence /
- phylogeny
Abstract:
| Citation: | DUAN Chunyan, WANG Xiaoling. Genetic characteristics of whole chloroplast genome in Prunus triloba ‘Multiplex’[J]. Journal of Zhejiang A&F University, 2024, 41(3): 577-585. DOI: 10.11833/j.issn.2095-0756.20230489

|


 DownLoad:
DownLoad: