





-
玉米Zea may是中国种植面积最大、产量最高的作物,既是保障国家粮食安全的核心基石,也是饲料生产和工业加工的关键原料[1]。生育期作为玉米生长周期的核心性状,直接决定品种的生态适应性和产量潜力,其中抽雄期、散粉期和吐丝期作为生育期的关键表征性状,既关乎品种熟期早晚,又与结实率及产量密切相关[2−3]。生育期受多基因协调调控,属于典型数量性状,在遗传分离群体中呈连续变异分布[4]。适宜生育期是玉米育种的核心目标之一,优化生育期对于提升玉米生产质量与效率具有重要意义。
数量性状位点(quantitative trait locus,QTL)定位是揭示复杂数量性状和鉴定候选基因的有效植物育种方法[5]。近年来,国内外学者利用不同玉米群体定位到了大量生育期相关数量性状位点。魏海忠等[6]研究表明:生育期相关QTL在染色体上呈“成簇”分布特征,且高贡献率QTL可调控多个相关性状。韩娅楠等[7]以自交系N6和BT-1为亲本构建重组自交系(recombinant inbred line,RIL)群体,在1号染色体umc1676~umc1590区间和2号染色体umc1422~umc1776区间鉴定到稳定调控抽雄期、吐丝期及散粉期的共同QTL位点。陈俊宇等[8]以玉米自交系LDC-1和TS501构建的186个重组自交系为材料,共检测到32个生育期相关的QTL。
浙江省地处东南沿海,独特的地理环境和气候条件孕育了丰富的玉米地方种质资源[9],但受国内外优良品种竞争冲击,且本地种质表型及基因型鉴定的精细度和系统性不足,致浙江地方种质的基因挖掘、分子育种及商业化开发利用程度低,遗传潜力未充分释放。当前,玉米生育期研究集中于QTL定位,深入挖掘生育期相关功能基因的研究仍较薄弱。本研究以浙江特色地方种质为核心,选取322份代表性玉米种质为材料,采用全基因组关联分析(genome-wide association study,GWAS)技术,结合单核苷酸多态性(single nucleotide polymorphism,SNP)标记技术,对玉米生育期性状开展系统解析,旨在揭示玉米种质生育期性状的遗传差异,挖掘生育期相关关键位点及候选基因,以期为玉米新品种精准选育提供理论支撑。
-
玉米供试材料为227份浙江省地方种质资源(自交4代)和145份国内外骨干自交系构成的群体[10]。该群体于2022—2023年分别种植于浙江省东阳市(29°28′N,120°33′E,海拔为161 m)和海宁市(30°15′N,120°41′E,海拔为5 m)。2022—2023年,受极端气候的影响,最终仅获322份玉米种质的完整生育期表型数据。
-
322份玉米供试材料于2022—2023年分别种植于浙江东阳和海宁,每个试点采用3次随机区组重复设计。小区种植2行,行距65 cm,株距33 cm,每行定植20株。2022年于浙江东阳繁种阶段,待玉米植株生长至五叶一心期时,选取322份种质的第三片功能叶单独采样,置于−80 ℃冰箱保存后,送至北京诺禾致源科技股份有限公司进行基因组重测序,用于构建基因型图谱。
-
抽雄期、散粉期及吐丝期的调查测定参照《玉米种质资源描述规范和数据标准》[11]执行,于各性状对应关键生育期开展观测。调查对象为各试验小区全部植株,判定标准如下:抽雄期记为小区内50%植株雄穗尖端露出顶叶的日期;散粉期记为小区内50%植株雄穗开始开花散粉的日期;吐丝期记为小区内50%植株雌穗花丝从苞叶吐出的日期。
-
采用Excel 2016和SPSS对不同环境下的玉米生育期表型数据进行统计分析。首先剔除异常值,随后计算最大值、最小值、平均值、标准差及变异系数,绘制群体频率分布及描述性图表,并对玉米生育期性状进行方差分析。利用R语言4.3.3分析群体生育期最佳线性无偏预测(best linear unbiased prediction,BLUP)。BLUP可整合多环境表型数据,剔除环境干扰效应,获得个体稳定遗传的表型值。
-
采用的基因型数据源自基因组变异数据库GVM,该数据库含22 642 752个SNP标记,平均密度为107 312个·Mb−1[10]。经322份自交系SNP位点筛选后,获得56 101个高质量SNP标记用于后续关联分析。采用Tassel 5.0软件的混合线性模型(mixed linear model,MLM),结合主成分分析(principal component analysis,PCA)和亲缘关系矩阵K,对多试点生育期表型数据及BLUP值开展关联分析;其中亲缘关系通过Tassel 5.0评估,PCA由R软件SNP Related包构建。以P<0.05为标准,设定动态阈值(0.05/n,n为SNP标记数)为标准,进行显著关联SNP位点鉴定,采用Excel 2016、R软件及ggplot2包进行数据分析和绘图,通过绘制曼哈顿图(GLM模型)验证并筛选最优模型。最终筛选出多试点及BLUP值均显著关联的SNP位点,通过匹配重叠位点鉴定稳定且环境依赖性低的核心位点。
-
由表1可见:2023年东阳的玉米抽雄期、散粉期及吐丝期均值均为最大,分别达74.74、78.17、79.22 d。2022年东阳玉米散粉期极差最大,为43.00 d,2022年海宁玉米吐丝期极差最小,为22.00 d。2022年海宁玉米抽雄期、散粉期及吐丝期的变异系数最大,均为0.10,2023年海宁玉米吐丝期变异系数最小,为0.06。
项目抽雄期/d 散粉期/d 吐丝期/d 2022年 2023年 2022年 2023年 2022年 2023年 东阳 海宁 东阳 海宁 东阳 海宁 东阳 海宁 东阳 海宁 东阳 海宁 最大值 73.00 71.00 94.00 84.00 80.00 74.00 97.00 86.00 68.00 74.00 99.00 86.00 最小值 34.00 46.00 55.00 49.00 37.00 49.00 59.00 51.00 36.00 52.00 60.00 52.00 极差 39.00 25.00 39.00 35.00 43.00 25.00 38.00 35.00 32.00 22.00 39.00 34.00 平均值 52.81 58.16 74.74 67.39 55.93 60.79 78.17 69.75 56.82 62.81 79.22 71.45 标准差 4.88 6.04 5.38 4.70 4.89 6.23 5.60 4.74 4.71 6.17 5.54 4.61 变异系数 0.09 0.10 0.07 0.07 0.09 0.10 0.07 0.07 0.08 0.10 0.07 0.06 Table 1. Descriptive statistical data of phenotypes for core traits of maize growth period across different experimental sites and years
通过对322份玉米供试材料4个试点的生育期核心性状表型数据联合分析(表2)显示:抽雄期平均值为63.49 d,极差为60.00,变异系数为0.16;散粉期平均值为66.41 d,极差为60.00,变异系数为0.16;吐丝期平均值为67.78 d,极差为63.00,变异系数为0.15。3个性状的平均值随抽雄期—散粉期—吐丝期依次递增,与玉米生育期自然发育进程一致;变异系数介于0.15~0.16,极差为60.00~63.00,整体变异跨度较大,表明供试群体生育期性状遗传多样性丰富,变异范围宽泛。
项目 抽雄期/d 散粉期/d 吐丝期/d 最大值 94.00 97.00 99.00 最小值 34.00 37.00 36.00 极差 60.00 60.00 63.00 平均值 63.49 66.41 67.78 标准差 10.38 10.53 10.50 变异系数 0.16 0.16 0.15 Table 2. Combined descriptive statistical analysis of core traits of maize growth period across multiple experimental sites and years
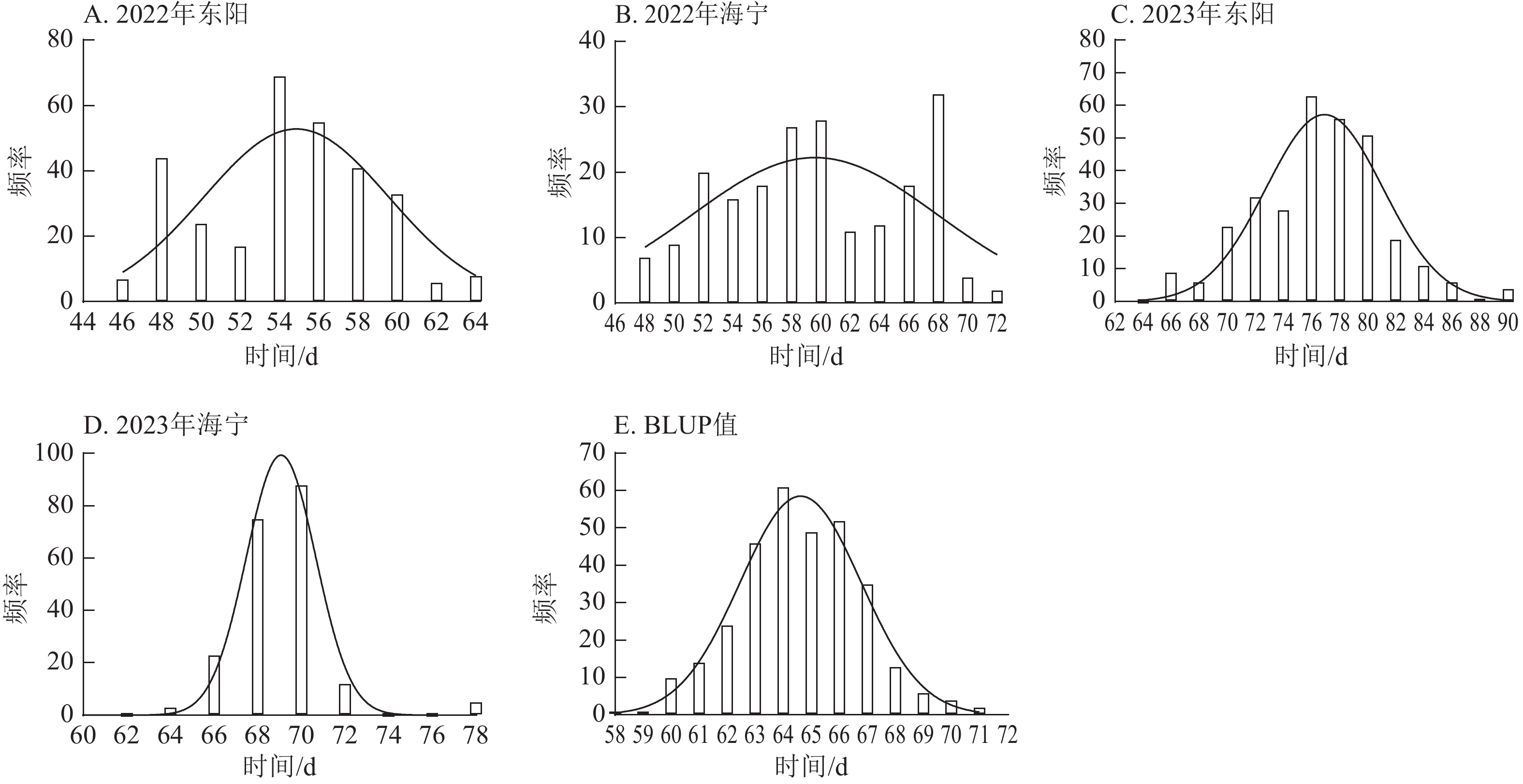
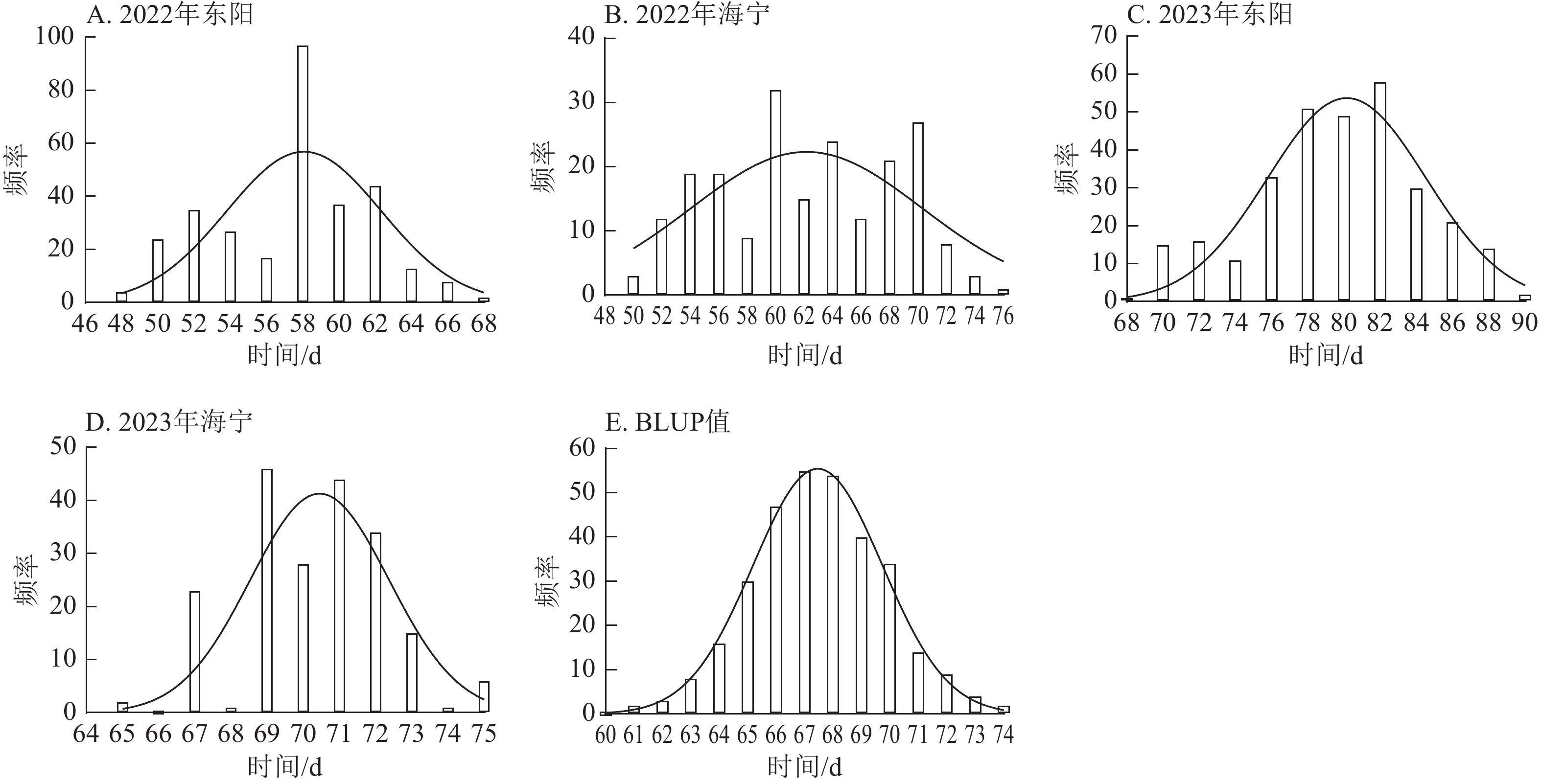
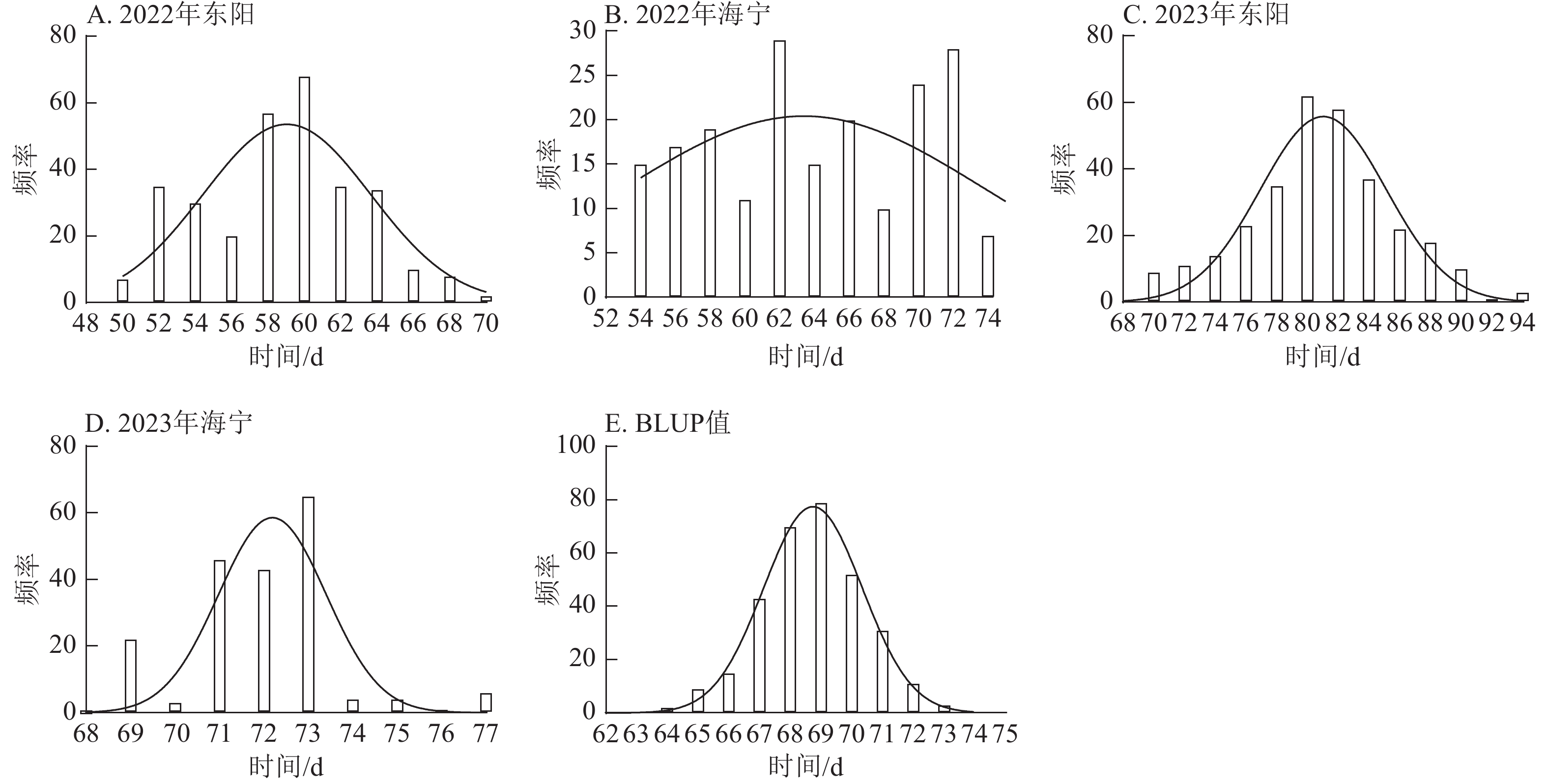
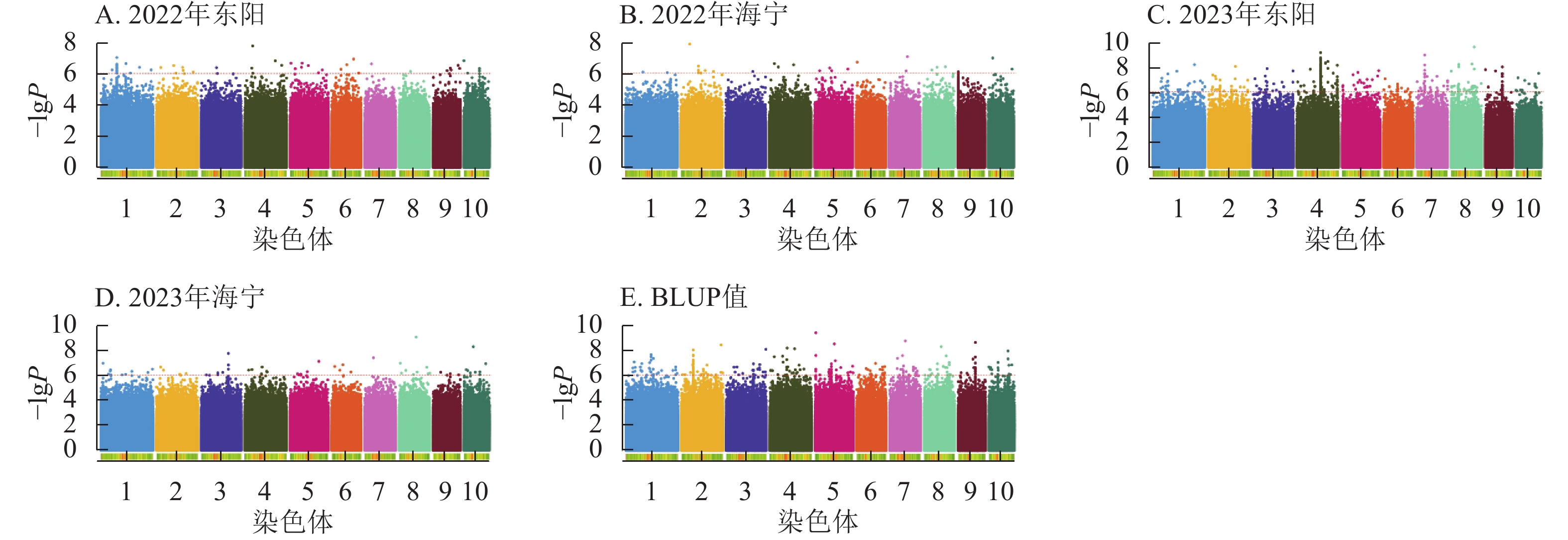
由图1~3表明:所有试点的生育期表型数据及性状BLUP值频率分布均呈正态分布,满足全基因组关联分析的基本要求。在抽雄期,2022年东阳试点主要分布于54和56 d,海宁试点主要分布于58、60和68 d;2023年东阳试点集中在76、78和80 d,海宁试点以68和70 d为主;BLUP值集中在63~66 d (图1)。在散粉期,2022年东阳试点集中于58 d,海宁试点分布于60和70 d;2023年东阳试点主要在78、80和82 d,海宁试点集中于70和72 d;BLUP值集中在66~68 d (图2)。在吐丝期,2022年东阳试点主要分布于58和60 d,海宁试点集中在62、70和72 d;2023年东阳试点以80~82 d为主,海宁试点集中于62、70和72 d;BLUP值集中在68和69 d (图3)。
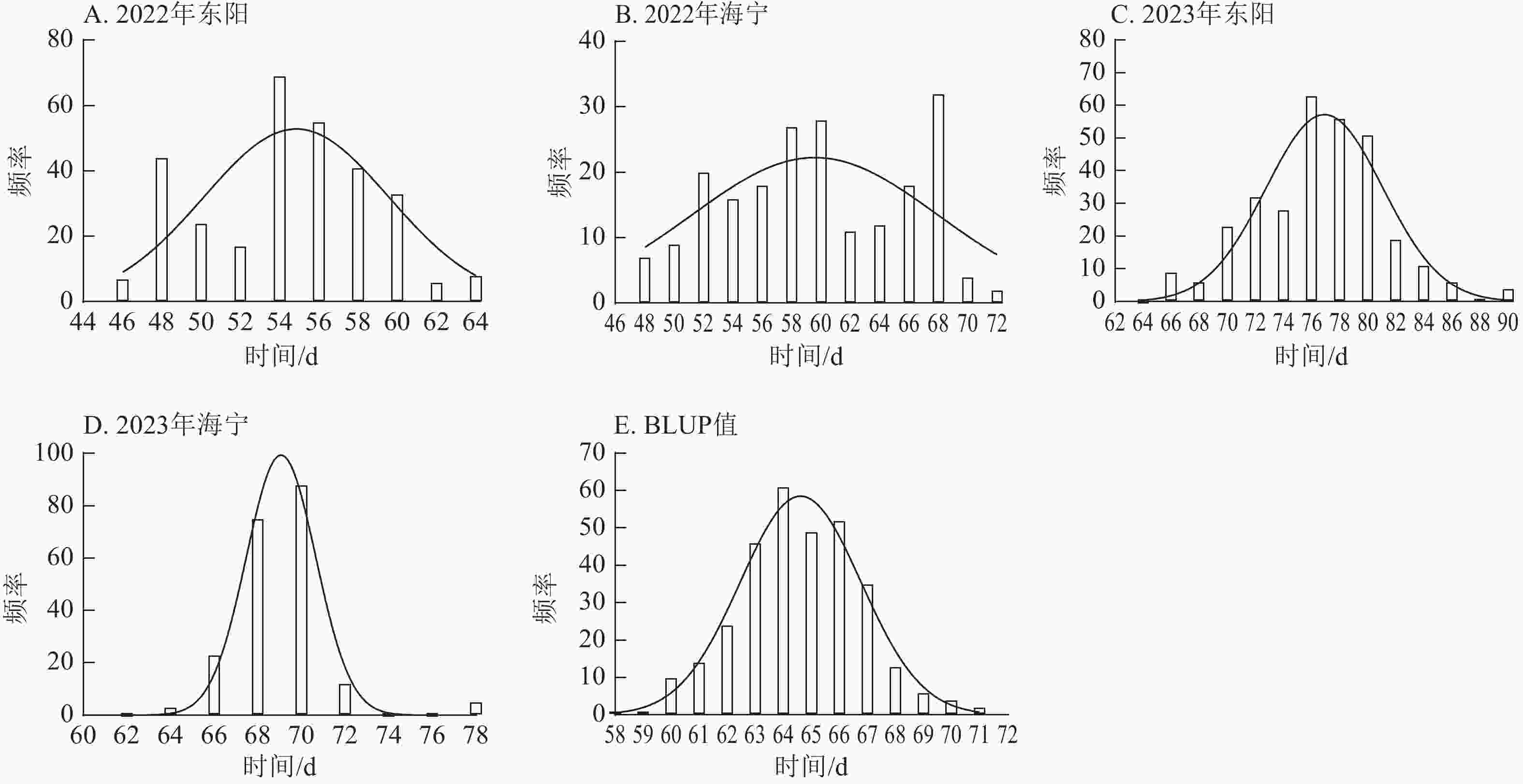
Figure 1. Frequency distribution of tasseling stage phenotypic data and BLUP values across different experimental sites
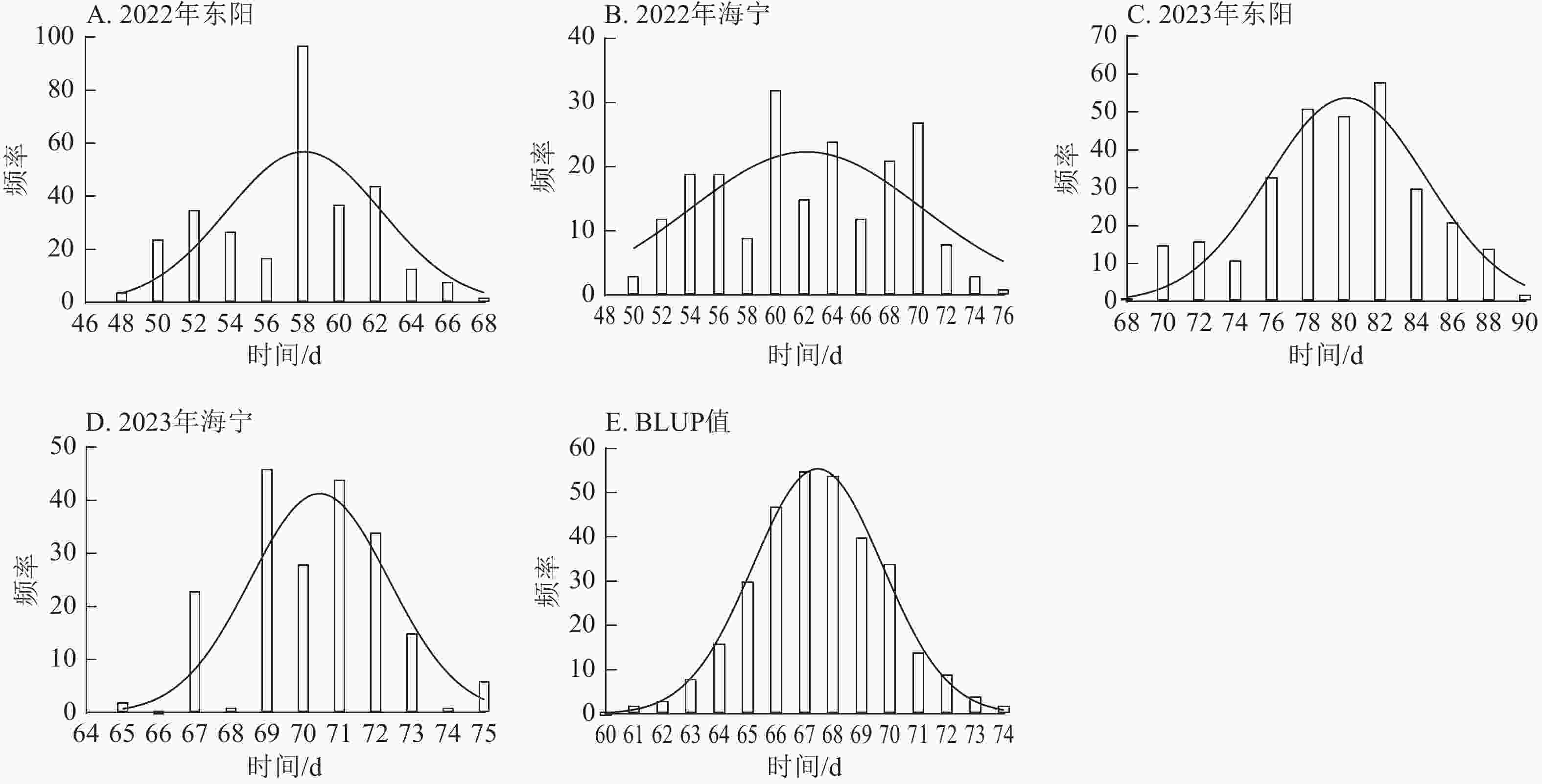
Figure 2. Frequency distribution of anthesis stage phenotypic data and BLUP values across different experimental sites
Figure 3. Frequency distribution of silking stage phenotypic data and BLUP values across different experimental sites
-
从表3可见:不同变异来源对各性状的影响存在差异。在抽雄期,品种效应极显著(P<0.001),品种×环境互作效应显著(P<0.01),而环境效应不显著;在散粉期,品种效应极显著(P<0.001),品种×环境互作效应极显著(P<0.001),环境效应不显著;在吐丝期,品种效应极显著(P<0.001),品种×环境互作效应极显著(P<0.001),环境效应显著(P<0.05)。综上表明供试材料3个生育期性状差异主要由遗传因素决定,且遗传效应受环境调控的程度因性状而异。
性状 变异来源 均方差 F P 抽雄期 品种 2469.700 58.167 <0.001*** 环境 74.292 1.750 0.155 品种×环境 127.541 3.004 0.002** 散粉期 品种 2473.762 51.700 <0.001*** 环境 109.079 2.280 0.078 品种×环境 158.809 3.319 <0.001*** 吐丝期 品种 2425.037 47.884 <0.001*** 环境 150.411 2.970 0.031* 品种×环境 172.716 3.410 <0.001*** 说明:*P<0.05;**P<0.01;***P<0.001。 Table 3. Results of combined analysis of variance for phenotypic data of core traits of maize growth period across multiple experimental sites
-
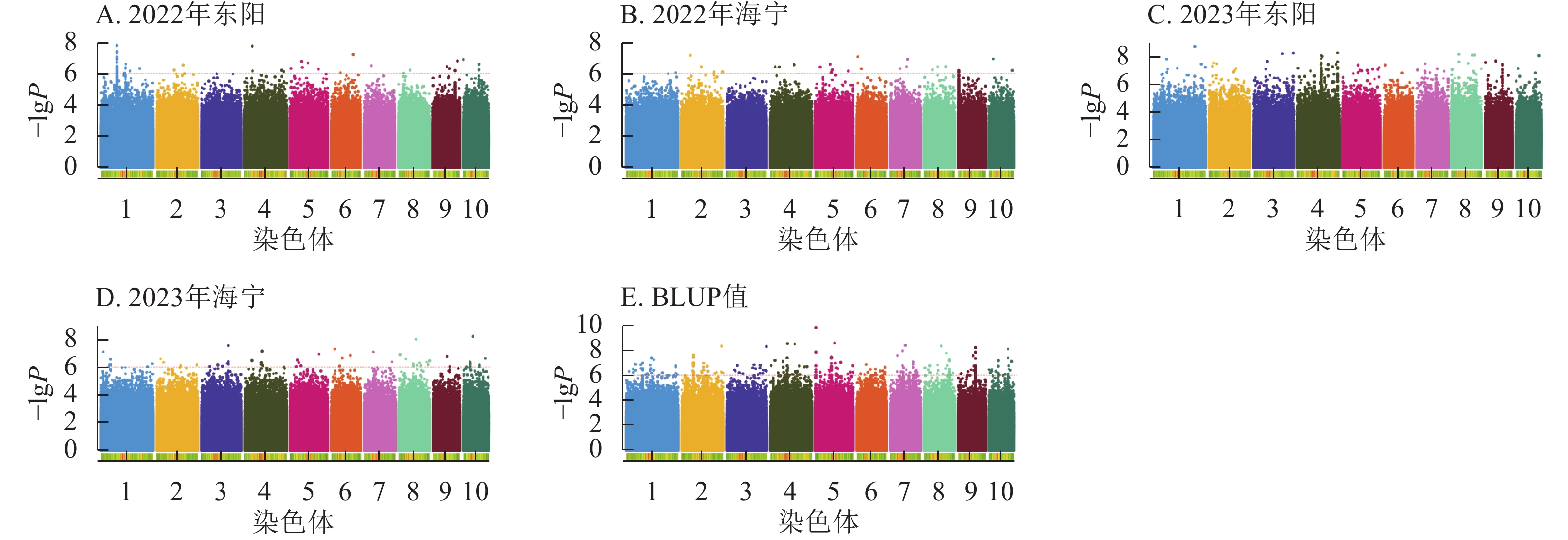
由图4显示:2022年东阳、海宁试点及2023年东阳、海宁试点分别鉴定到抽雄期显著关联SNP位点61、27、281、57个,平均表型变异解释率依次为7.87%、7.94%、8.01%、7.94%;基于BLUP值的分析共检测到241个显著位点,平均表型变异解释率为7.97%。
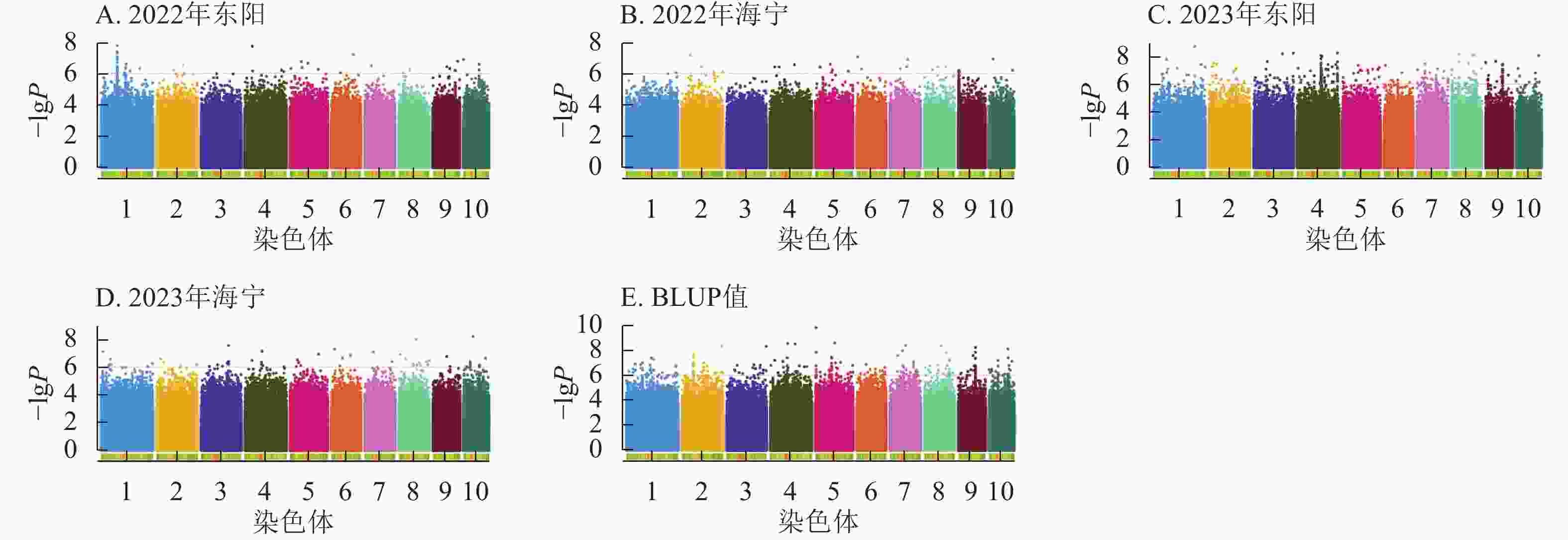
Figure 4. Manhattan plots of significantly associated SNPs for tasseling stage trait across multiple experimental sites
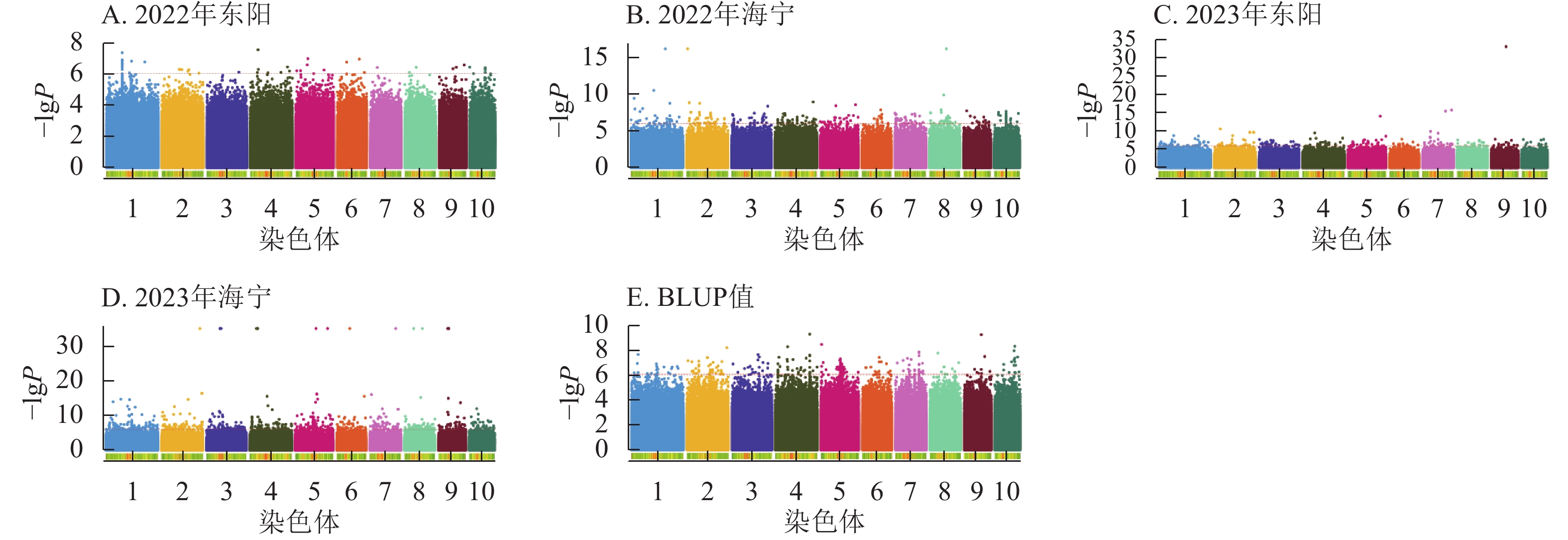
图5显示:2022年东阳、海宁分别检测到51、26个散粉期性状显著位点,平均表型变异解释率分别为7.70%、7.89%;2023年东阳、海宁分别检测到424、58个显著位点,平均表型变异解释率分别为8.12%、7.97%;基于BLUP值分析共检测到220个显著位点,平均表型变异解释率为7.98%。
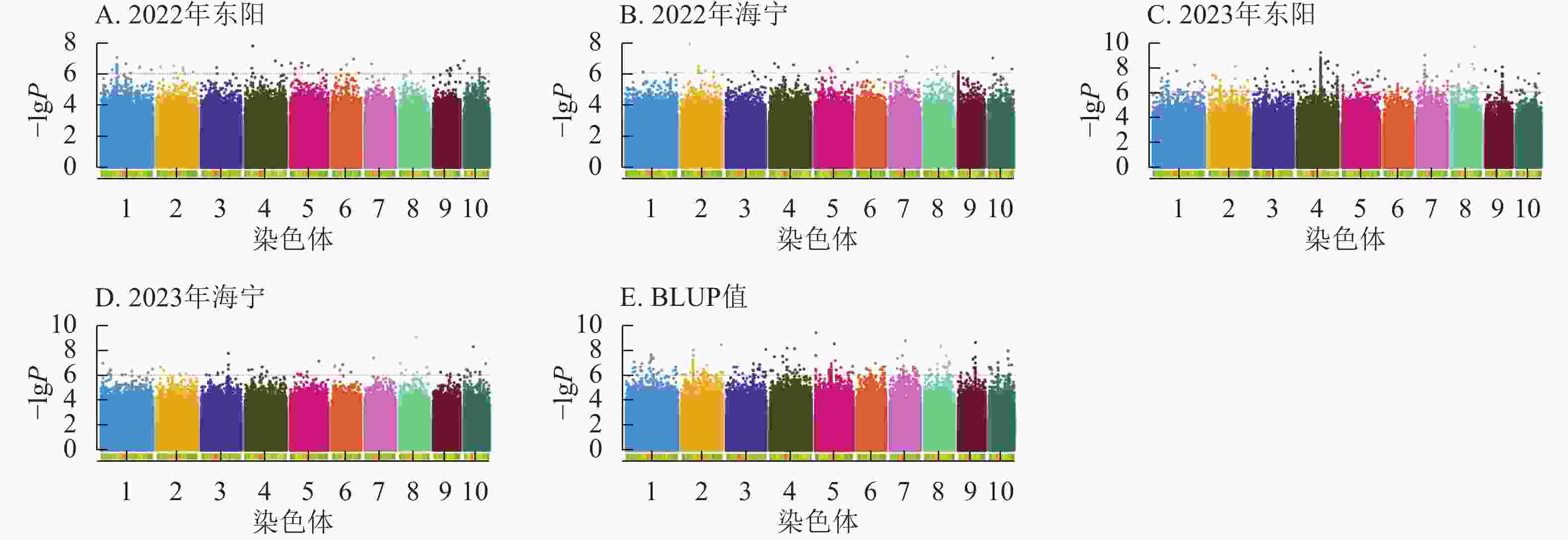
Figure 5. Manhattan plots of significantly associated SNPs for anthesis stage trait across multiple experimental sites
图6显示:2022年东阳、海宁分别检测到47、277个吐丝期性状显著位点,平均表型变异解释率为7.79%、8.39%;2023年东阳、海宁分别检测到212、1 169个显著位点,平均表型变异解释率为8.54%、9.13%;基于BLUP值分析共检测到243个显著位点,平均表型变异解释率为7.95%。
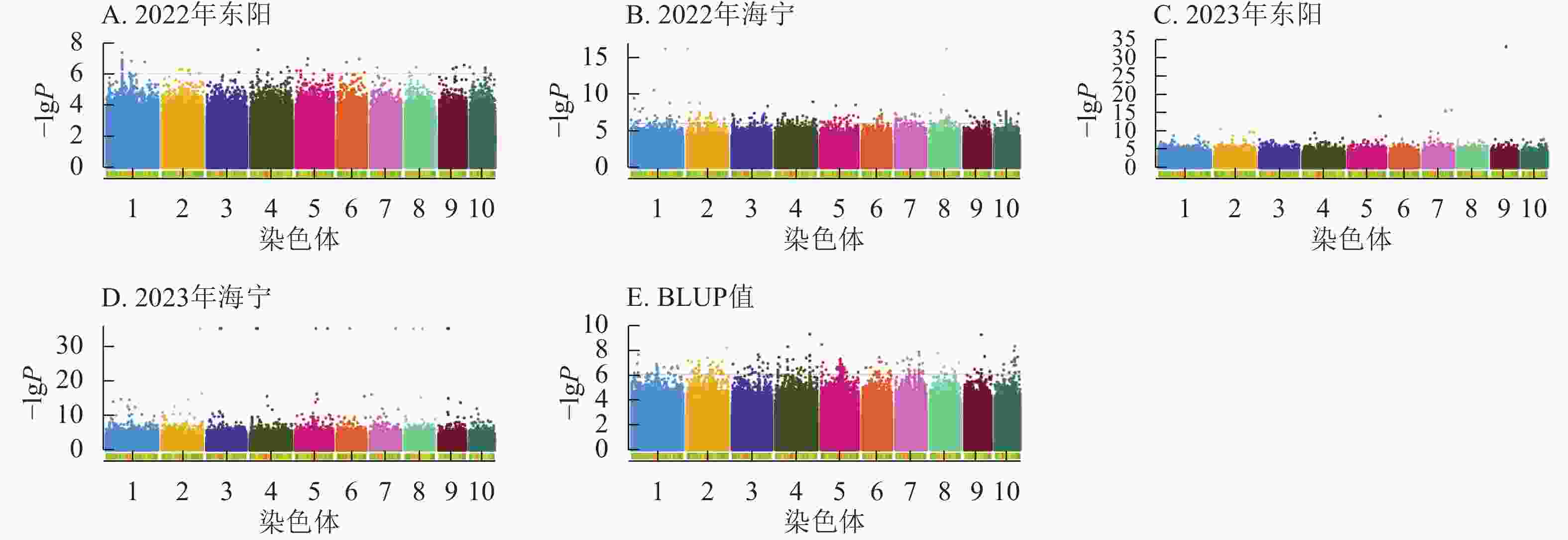
Figure 6. Manhattan plots of significantly associated SNPs for silking stage trait across multiple experimental sites
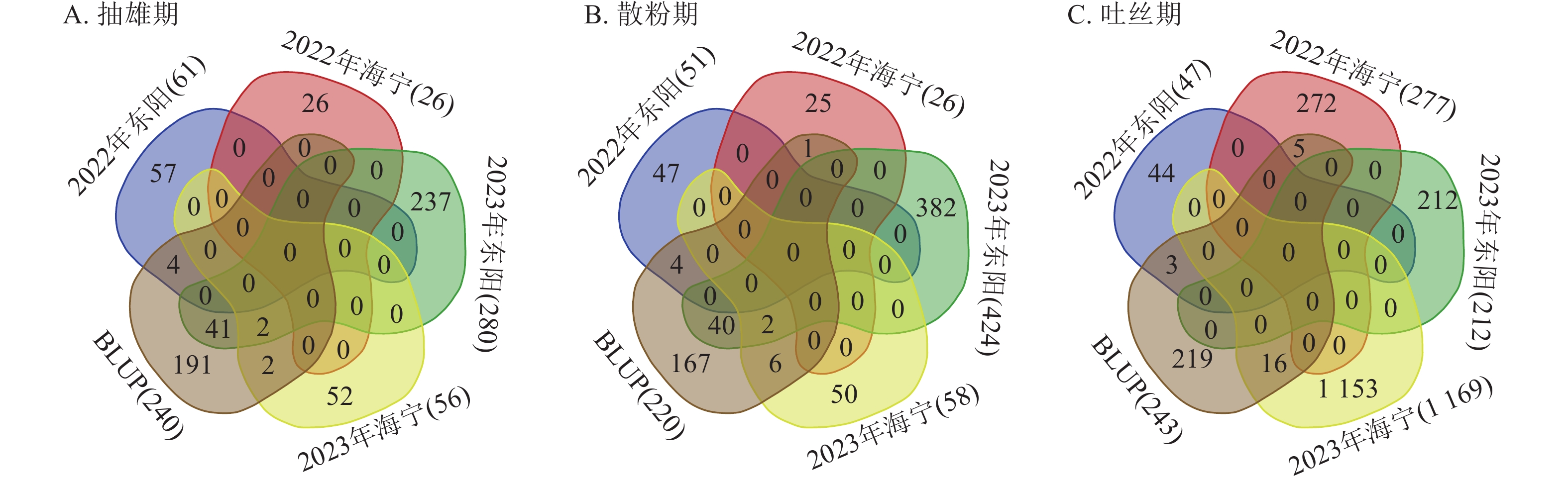
对各试点生育期性状关联的显著SNP位点进行重叠性分析(图7)发现:抽雄期共检测到49个重叠显著SNP位点,主要分布于1、3、4、5、6、7、8、9、10号染色体,表型变异解释率为7.26%~12.07%,其中5号染色体上的S5_4607205表型变异解释率最高(12.07%)。这些位点附近共鉴定出43个候选基因,分布于1、3、4、5、7、8、9、10号染色体,含1个位于基因内部的SNP位点。功能注释显示:候选基因主要参与蛋白翻译、膜运输及细胞周期调控等生物学过程,其中S10_11955883、S3_160561791、S3_196947385、S4_102188497、S4_79045396、S9_17461941附近的Zm00001d023611、Zm00001d042314、Zm00001d043335、Zm00001d050577、Zm00001d050293、Zm00001d045268已有相关研究报道[12−17]。
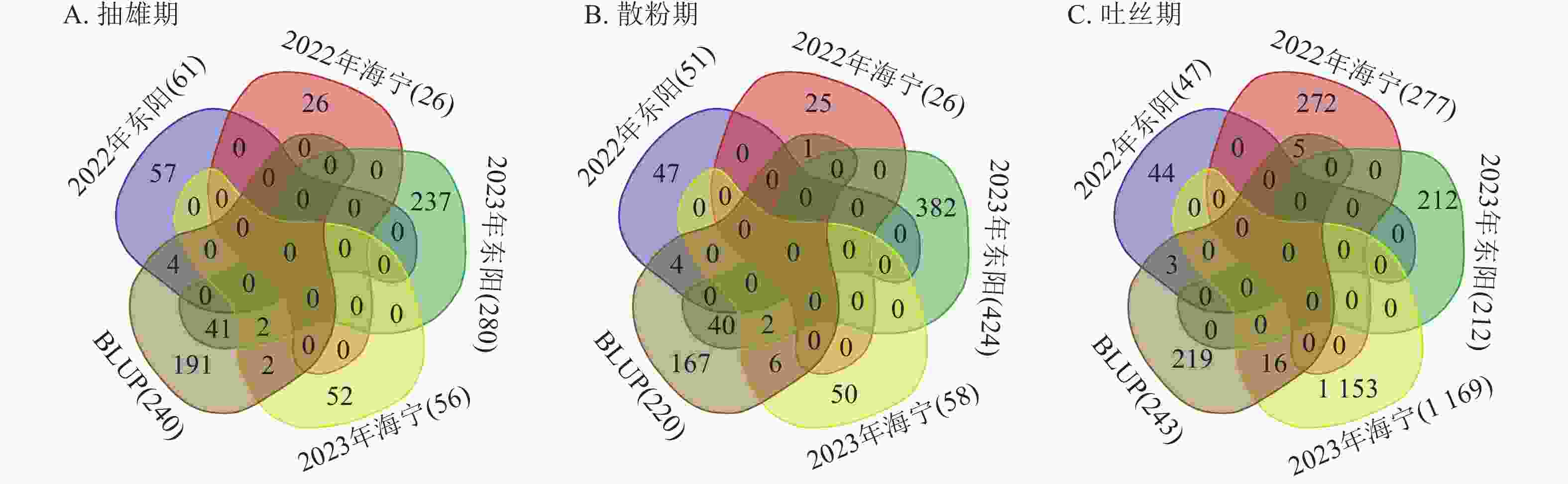
Figure 7. Statistics of the number of overlapping significantly associated SNP loci for core growth period traits across multiple experimental sites
散粉期共检测到53个重叠显著SNP位点,主要分布于1、2、3、4、5、8、9、10号染色体,表型变异解释率为7.25%~11.55%,其中S5_
4607205 表型变异解释率最高(11.55%)。这些位点附近共鉴定出38个候选基因,分布于1、2、3、4、5、7、8、9、10号染色体,含有4个位于基因内部的SNP位点。功能注释表明:候选基因主要涉及细胞离子平衡、信号传导及催化作用等生物学过程,其中S10_11955883 、S4_79045396 附近的Zm00001d023611、Zm00001d050293与抽雄期候选基因重叠,且已有研究报道[12, 15],提示散粉期与抽雄期可能共享部分调控机制。吐丝期共检测到24个重叠显著SNP位点,主要分布于1、2、4、5、7、8号染色体,表型变异解释率为7.26%~11.35%,其中4号染色体上的S4_
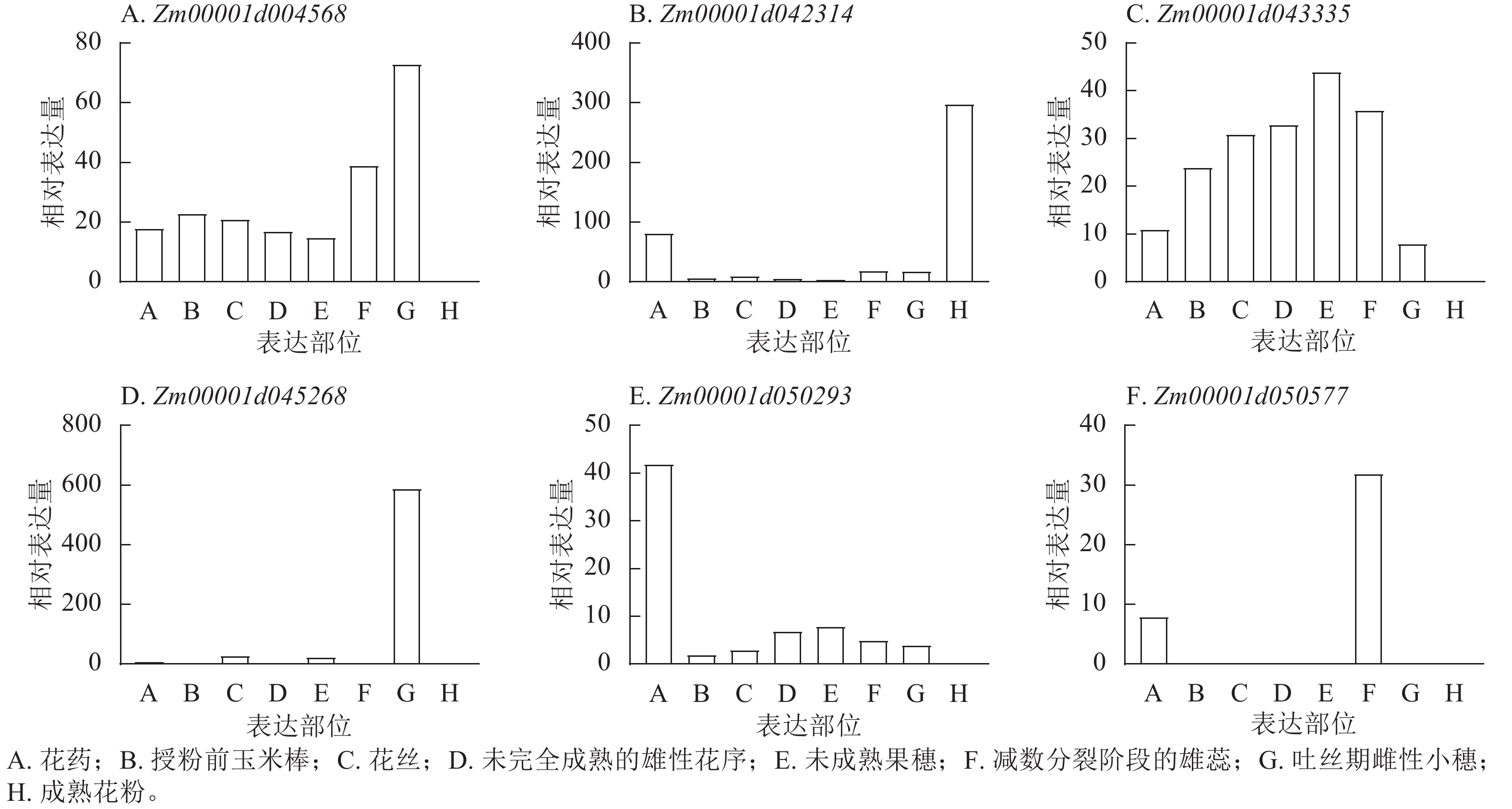
201865940 表型变异解释率最高(11.35%)。这些位点附近共鉴定出9个候选基因,分布于2、4、5、7、8号染色体,功能注释显示:这些候选基因主要参与植物生长控制与信号传导等生物学过程,其中S3_118445252 、S7_81942899 附近的Zm00001d004568、Zm00001d004571、Zm00001d019983已有相关研究报道[18−19]。通过公共数据库查询抽雄期候选基因的组织表达谱发现:Zm00001d042314、Zm00001d043335、Zm00001d050577、Zm00001d050293、Zm00001d045268、Zm00001d004568等基因在生育期相关组织中表达量较高,提示其可能参与玉米生育期性状的调控(图8)。
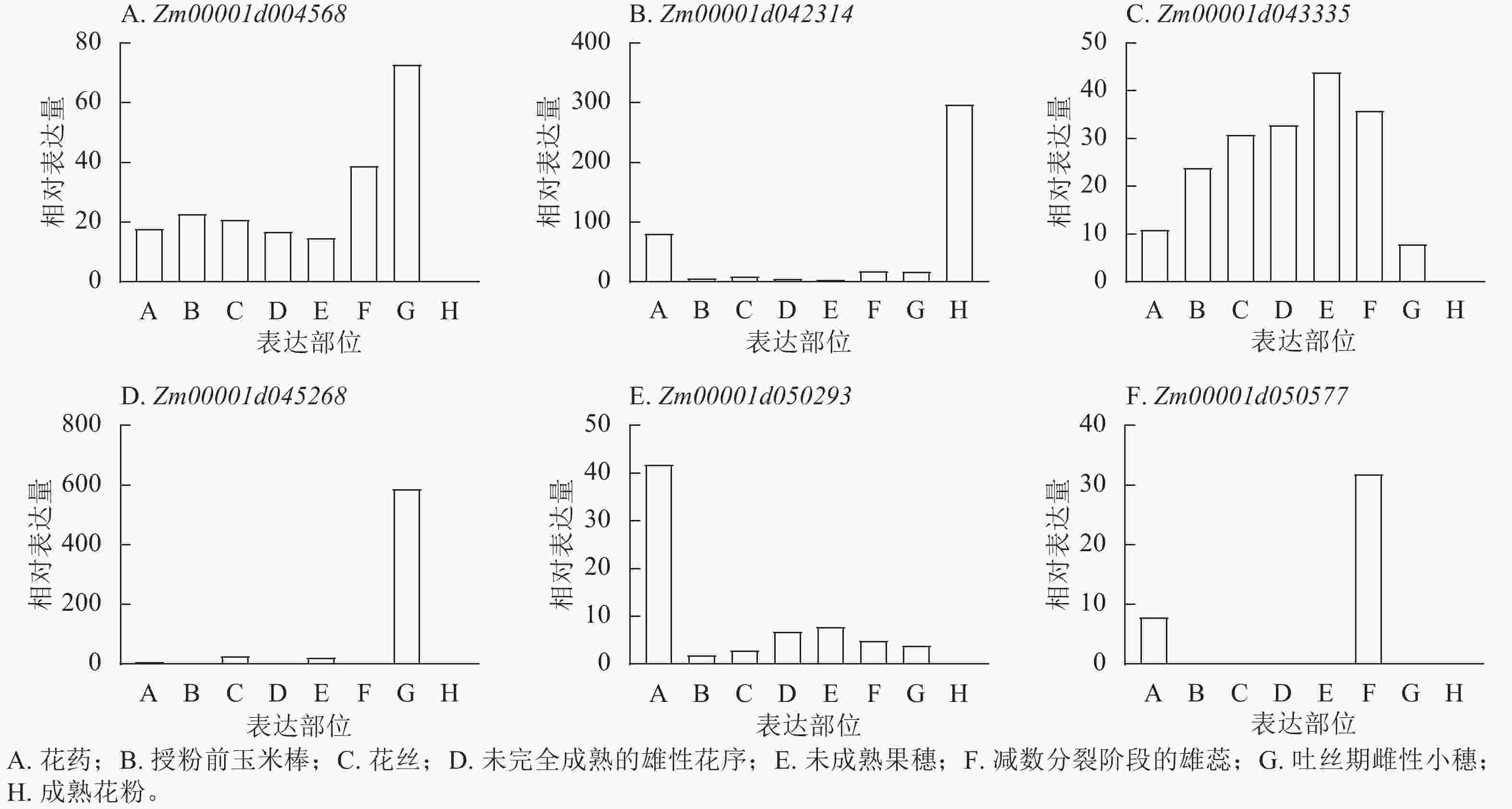
Figure 8. Analysis of relative expression profiles of candidate genes for maize growth period in different tissue parts
-
李玉玲等[20]以普通玉米自交系丹232和爆裂玉米自交系N04为亲本构建家系群体,在1号染色体umc1403~phi001区域检测到控制抽雄期的QTL,在1号染色体bnlg1429~umc1403区域检测到抽雄期和吐丝期的QTL,在5号染色体bhlg565~umc1389区域同时检测到控制抽雄期和散粉期的QTL。本研究通过全基因组关联分析,在1号染色体umc1403~phi001区间内定位到S1_
34410222 等14个SNP位点,在bnlg1429~umc1403区域定位到S1_17532310 等22个SNP位点,在5号染色体bhlg565~umc1389区域定位到S5_143819515 等66个SNP位点。韩娅楠等[7]以自交系N6和BT-1为亲本构建重组自交系群体,发现1号染色体umc1676~umc1590区域和2号染色体umc1422~umc1776区域存在共同控制抽雄期、吐丝期和散粉期的稳定QTL。本研究在上述2个区域分别定位到与生育期相关的S1_151244200 等47个SNP位点和S2_1015268 等15个SNP位点,这与前人研究结果高度一致[7, 20],因此证实了本研究关联SNP位点的可靠性。此外,本研究还新增定位相关SNP位点抽雄期49个、散粉期53个、吐丝期24个,为玉米生育期性状的遗传调控研究提供了重要补充。开花期是影响玉米生长发育的关键时期。马拴红等[21]研究发现:Zm00001d034036、Zm00001d020364、Zm00001d047269等早花蛋白基因受到ARF、MYB和NAC转录因子调控,通过下游基因表达调控玉米开花期。本研究中,SNP位点S1_
280457376 位于Zm00001d034036附近,S7_109168545 、S7_109927283 位于Zm00001d020364附近。党昆泰[22]研究表明:ZmHB53、ZmMADS6、ZmMYB43、ZmMYB93、ZmCCD7等基因与玉米开花时间、结实以及激素信号响应密切相关,是受IAA29-ARF蛋白质复合物影响的关键下游基因。本研究定位到的S6_140804982 、S6_140809095 位点位于ZmMYB93附近,进一步验证了本研究生育期相关候选基因的可靠性。对多试点抽雄期、散粉期、吐丝期重叠位点的分析显示:42个SNP位点在3个性状中共同存在,提示可能存在同时调控这3个生育期性状的候选基因。结合全基因组关联分析筛选的SNP重叠位点、Maize GDB数据库的功能注释及组织表达谱结果,本研究筛选到5个抽雄期候选基因(Zm00001d042314、Zm00001d043335、Zm00001d050577、Zm00001d050293、Zm00001d045268),1个散粉期候选基因(Zm00001d050293),1个吐丝期候选基因(Zm00001d004568)。郭爽等[23]预测发现:12个开花期相关候选基因主要参与编码蛋白质丝氨酸/苏氨酸激酶活性,沈辰[24]研究发现:丝氨酸/苏氨酸蛋白磷酸酶基因都与山核桃Carya cathayensis雌雄花分化有关,而本研究初步筛选的散粉期候选基因Zm00001d005501编码蛋白与之功能一致,且该基因在玉米中尚未见报道,提示其可能为调控玉米生育期的新候选基因。
YI等[25]发现经典H3基因ZmH3-a和ZmH3-b在玉米穗期高表达。本研究中,Zm00001d042314编码含胱硫醚β合酶(CBS)结构域的蛋白,FANG等[26]及KUSHWAHA等[27]研究表明:含CBS结构域的蛋白质可能参与生殖发育调控,虽其在玉米中的功能尚未明确,但推测其可能通过该结构域影响植株生长发育,进而调控生育期。Zm00001d043335在生育期相关组织中高表达,提示其可能为调控生育期的关键基因。Zm00001d050577编码糖转运蛋白15a,且在生育期相关组织中高表达;GENDRON等[28]研究发现:拟南芥Arabidopsis thaliana在诱导性长日照条件下,茎尖附近韧皮部中的蔗糖含量会出现短暂上升[29],推测Zm00001d050577可能通过参与糖转运过程影响生育期调控。Zm00001d050293编码糖-海藻糖-6-磷酸(T6P),作为重要信号分子,T6P水平与蔗糖含量在光暗周期中密切关联[30]。在拟南芥中,T6P合成减少会导致FT基因表达,过量合成则会促进短日照和长日照条件下的开花[31],表明T6P可介导碳水化合物状态与光周期性开花的关联,为光合周期与开花调控搭建桥梁。据此推测,Zm00001d050293可能通过调控光暗周期中蔗糖与T6P的关联,影响玉米生育进程。Zm00001d045268在生育期相关组织中高表达,提示其可能参与玉米生育期调控。此外,本研究筛选的候选基因还包括编码膜丝氨酸/苏氨酸激酶BRI1蛋白、茉莉酸钠诱导蛋白的基因。当油菜素类固醇(BR)被跨膜丝氨酸/苏氨酸激酶BRI1感知时,BRI1被激活并通过下游蛋白磷酸化转导信号[32];油菜素类固醇不敏感2 (BIN2)作为油菜素类固醇信号通路的负调节因子,在无油菜素类固醇存在时可磷酸化转录因子BZR1和BES1,抑制其活性[33−37],进而影响植物生长发育。
玉米ZmDWARF8等基因通过调控植株从营养生长到生殖生长的转换过程,影响开花时期[38−39],ZCN8基因作为玉米光周期调节途径的关键基因,对开花过程具有促进作用[40−42],本研究表明:不同玉米品种间生育期存在显著差异,其中普通玉米品种的极差最大,可能与该类品种样本量较大有关;不同试点间生育期亦存在明显差异,如2023年浙江东阳与海宁试点的散粉期平均值相差8.42 d。LI等[43]发现:玉米抽穗发育的分子调控与早期营养生长阶段的水分亏缺胁迫有关;刘月娥[44]研究表明:环境对生育期相关性状的影响显著,本研究方差分析结果亦显示:环境间效应及环境与品种的互作效应均达到显著水平。2022年2个试点的生育期较2023年更短,推测与2022年播种期较晚,生育期内总体气温较高有关,进一步证实生育期与气温存在密切关联。
-
本研究新鉴定出抽雄期、散粉期、吐丝期多试点重叠显著SNP位点分别为49、53、24个,筛选到5个抽雄期、1个散粉期及1个吐丝期候选基因。其中,Zm00001d042314可能参与生殖发育调控生育期,Zm00001d043335或通过调控叶绿体影响光合途径进而作用于生育期,Zm00001d050293可通过关联光暗周期中蔗糖水平介导光合周期与开花的联系;这些候选基因分别在成熟花粉、未成熟果穗、未授粉叶片及吐丝期雌性小穗等生育期相关组织中高表达,与性状表现高度契合,为玉米生育期性状相关基因挖掘提供了重要参考。
Genome-wide association study of growth traits in 322 maize germplasms
doi: 10.11833/j.issn.2095-0756.20250210
- Received Date: 2025-03-19
- Accepted Date: 2025-10-31
- Rev Recd Date: 2025-10-29
- Available Online: 2026-04-02
- Publish Date: 2026-04-02
-
Key words:
- maize (Zea mays) /
- growth period /
- genome-wide association study (GWAS) /
- candidate genes
Abstract:
| Citation: | CHEN Yong, LUO Yao, SHEN Xiaobin, et al. Genome-wide association study of growth traits in 322 maize germplasms[J]. Journal of Zhejiang A&F University, 2026, 43(2): 320−330 doi: 10.11833/j.issn.2095-0756.20250210

|
 DownLoad:
DownLoad: