







-
转座子(transposable elements, TEs)是一种自私的基因组“寄生虫”,能够增加拷贝数并改变其在宿主基因组中的位置[1]。转座子由于具有突变的可能,会对邻近基因表达产生潜在的危害,并导致染色体重排,对基因组的稳定性构成威胁[2-4]。转座子根据其转座中间产物被分为2类,其中Ⅰ类转座子包括复制黏贴式反转录转座子(retrotransposons, REs)[5],Ⅱ类转座子包括剪切黏贴式转座子[6]。Ⅰ类转座子根据其内部的结构可以分为LTR类(long terminal repeat retrotransposons)[6]、DIRS类(dictyostelium intermediate repeat sequence elements)[7]、PLE类(penelope-like elements)[8]、LINE类(long interspersed nuclear elements)[9]、SINE类(short interspersed nuclear elements)[10]。其中LTR反转录转座子是至今为止研究最多的一类[6]。LTR反转录转座子具有4个结构特点。第一,在序列两端有1对靶位点重复序列(target site repeats,TSD),约4~6 bp[11];第二,5′端和3′端有1对长为几十到几千bp不等高度相似的长末端重复序列[12];第三,LTR主要包括GAG衣壳蛋白编码区和POL多蛋白编码区(polyprotien),其中POL包括RH核糖核苷酸酶(ribonuclease h,RNaseH)、RT反转录酶(reverse transcriptase)、INT整合酶(integrase)和AP蛋白酶(aspartic proteinase)。还有一些Retrovirus和ENV超家族(superfamily)的LTR含有ENV序列(envelope protein, EN或ENV)[13];第四,在5′端附近有1个引物结合位点(primer binding site,PBS),可调控其基因组RNA反转录所必需的tRNA引物,它以染色体外线性DNA(extrachromosomal linear DNA, eclDNA)的形式产生LTR反转录转座子生命周期中间体。在3′端附近有1个富嘌呤位点(poly purine trait,PPT)[14],协助反转录的完成。根据LTR反转录转座子开放阅读框(open reading frames,ORF)的完整性分为自主LTR反转录转座子和非自主LTR反转录转座子[15]。自主LTR反转录转座子又可以根据POL中RT、INT和RH编码序列的排列方式,分为Ty1-copia 超家族(5′-INT-RT-RH-3′)和Ty3-gypsy超家族(5′-RH-RH-INT-3′)[16]。根据每个超家族的序列同源性(80-80-80的分类规则[17])可以分为不同的家族。根据LTR反转录转座子的同源性、蛋白结构、进化关系可以划分为不同的谱系,如梨Pyrus基因组中被划分为Ale、Ivana、Bianca、Angela、Tar、Tat、Athila、Renia、Crm、Galadriel、Tekay等11个谱系[18]。LTR反转录转座子的活性包括转录活性和转座活性。转录是转座的第1步,许多LTR反转录转座子在植物杂交、多倍体化或在环境挑战下发生去甲基化,被转录激活[19]。转座活性不仅包括转录活性,还受转录后调控,如植物基因组为了抑制转座子的活性通过转录基因沉默(transcriptional gene silencing, TGS)机制抑制它们的能力[20]。如果TGS得到缓解,则受21~22个核苷酸作用的转录后基因沉默机制(post-transcriptional gene silencing, PTGS)会将靶向转座子转录物进行降解[21],所以在实验条件下LTR反转录转座子很难被转座激活。在整个植物王国中LTR反转录转座子的进化特别成功,不断复制转座,导致基因组大小增加,基因组尺寸产生差异。在被子植物基因组之间由于几个LTR反转录转座子家族的扩增,产生一些巨大的基因组。例如2 400 Mb玉米Zea mays[22]和400 Mb水稻Oryza sativa[23]的基因组中LTR反转录转座子家族数相同,但是玉米基因组中5个谱系的LTR反转录转座子拷贝数较高。即使是亲缘关系很近的品种,LTR反转录转座子也会促使它们的基因结构产生巨大差异。如玉米与大刍草Zea mexicana是近亲,但是大刍草的基因组比玉米大1倍[24]。LTR反转录转座子在植物基因组中处于动态变化的过程,不仅会扩增,也会丢失,不平衡重组(illegitimate recombination)和非法重组(unequal recombination)活动就是丢失的主要原因[25-26]。不平衡重组和非法重组的产物主要包括含有TSD位点的solo LTR,不含TSD位点的Truncated LTR[27]。预测LTR反转录转座子可以通过4种方式[28]:比较基因组法(comparative genomic methods)[29]、重复序列从头算起法(de novo repeat discovery)[30]、同源比对法(homology-based methods)[31]、基于结构预测法(structure-based methods)。基于结构预测法是通过LTR反转录转座子的序列结构和转座机制分析来捕获,如LTR_STRUC、LTR_FINDER、LTRharvest、LTR_par、LTR_Rho等[32]。毛竹Phyllostachys edulis具有较高的经济和生态价值,其种植面积在中国的竹子总种植面积(443 万hm2)中占73.76%[33]。2018年第2版毛竹基因组的公布[34]为深入分析毛竹基因组中LTR反转座子提供了良好的条件。本研究运用了LTRharvest的方法[35],对第2版毛竹基因组中的LTR反转录转座进行预测,并对LTR反转录转座子的结构、在基因组中的分布特征、插入时间等进行系统分析,以期能了解毛竹LTR反转录转座子对基因组的影响。
HTML
-
第2版毛竹基因组序列、基因组注释文件来自毛竹基因组数据库[34]。先利用LTRharvest软件[35]预测毛竹基因组LTR反转录转座子,利用cd-hit根据80-80-80的分类规则[17]进行序列同源性聚类,然后利用LTRdigestion[36]注释各个LTR反转录转座子结构域。接着根据LTR反转录转座子的POL编码区RT、INT和RH的顺序将其分为Ty1-Copia(INT-RT-RH)和Ty3-Gypsy (RT-RH-INT)2个超家族[16]。根据序列中的GAG和POL编码区与Gypsy Database 2.0网站中(http://gydb.org/index.php/Phylogeny: POL_LTR_retroelements)植物典型的Tork、Reftrofit、Sire、Oryco、Del、Reina、Crm、Tat、Galadriel和Athila 谱系对应编码区的同源性,进一步将2个超家族分为10个谱系[35]。最后利用RepeatMakser软件分析LTR反转录转座子在毛竹基因组中的含量[31]。
-
使用MA等[37]对每条结构完整的毛竹LTR反转录转座子的插入时间进行计算:①对每条结构完整的LTR反转录转座子的两端LTR序列利用MUSCLE软件[38](使用默认参数)进行比对;②利用JUCKES-CANTOR的方法[39]计算碱基突变频率(K);③利用公式T=K/(2r)计算插入时间,T表示时间,r表示生物钟,r=1.3×10−8 bp·a−1[37]。
1.1. 毛竹LTR反转录转座子的鉴定与分类
1.2. 毛竹LTR反转录转座子插入时间计算
-
由表1所示:得到1 014 565条LTR反转录转座子,占整个毛竹基因组的54.97%。毛竹中LTR反转录转座子比例与其他基因组相比,低于玉米基因组的70.1%[22],相近于高粱Sorghum bicolor基因组的55%[40],远高于水稻基因组的26%[23]。其中两端具有完整LTR序列,编码结构域完整的LTR反转录转座子(full-length LTR)有7 731条,两端具有完整LTR序列,编码结构域不完整的LTR反转录转座子(solo LTR)有13 656条(其余不含TSD位点的LTR反转录转座子忽略不计)。然后按照WICKER等[16]提出的真核生物转座子的分类方法,将blastn(all-vs-all)的方法和80-80-80的规则相结合,对7 731条完整的LTR反转录转座子进行分类,共分为1 562个家族。
超家族 谱系 家族a 结构 数量/个 全长/bp 百分比b/% Ty1-copia Tork 236 GAG-PR-INT-RT-RH 145 708 124 219 995 6.51 Retrofit 342 GAG-PR-INT-RT-RH 41 965 43 615 815 2.29 Sire 136 GAG-PR-INT-RT-RH-ENV 223 386 210 097 734 11.01 Oryco 105 GAG-PR-INT-RT-RH 22 078 22 854 591 1.20 合计 819 433 137 400 788 135 21.01 Ty3-gypsy Del 207 GAG-PR-RT-RH-INT-CHR 295 222 334 005 916 17.51 Reina 249 GAG-PR-RT-RH-INT-CHR 27 803 39 235 939 2.06 Crm 47 GAG-PR-RT-RH-INT 40 781 44 298 955 2.32 Tat 238 GAG-PR-RT-RH-INT 217 288 230 055 053 12.06 Galadriel 1 GAG-PR-RT-RH-INT-CHR 23 51 248 0.00 Athila 1 GAG-PR-RT-RH-INT-ENV 311 257 970 0.01 合计 743 581 428 647 905 081 33.96 总计 1 562 1 014 565 1 048 693 216 54.97 说明:a表示每个谱系的数量;b表示在毛竹基因组中LTR反转录转座子所占的比例 Table 1. Classification of LTR retrotransposons superfamily of moso bamboo genome
毛竹LTR反转录转座子分为Ty1-copia和Typ3-gypsy 2个超家族,在1 562个LTR反转录转座子家族中有819个家族属于Ty1-Copia 超家族,共包括433 137条序列,长度为400 788 135 bp,占毛竹基因组的21.01%。743个家族属于Ty3-Gypsy超家族,共包括581 429条序列,长度为647 905 081 bp,占毛竹基因组的33.96%(表2)。Ty3-gypsy与Ty1-copia数量之比为1.3∶1.0,低于大豆Glycine max(1.4∶1.0)[19]和玉米(1.6∶1.0)[22],远低于水稻(4.9∶1.0)[41]和高粱(3.7∶1.0)[40],但远高于苜蓿Medicago sativa(0.3∶1.0)[42]。
谱系 家族a 百分比c/% 全长LTRd Solo LTRe 全长LTR/Solo LTR 全长 LTR+Solo LTR 百分比f/% Tork 236 28.82 1 169 1 492 1.28 2 661 28.76 Retrofit 342 41.76 302 1 158 3.83 1 460 15.78 Sire 136 16.61 464 3 139 6.77 3 603 38.95 Oryco 105 12.82 521 1 006 1.93 1 527 16.51 Ty1-copia 819 100.00 2 456 6 795 2.77 9 251 100.00 Del 207 27.86 2 102 2 992 1.42 5 094 41.97 Reina 249 33.51 495 1 245 2.52 1 740 14.34 Crm 47 6.33 510 1 352 2.65 1 862 15.34 Tat 238 32.03 2 168 1 251 0.58 3 419 28.17 Galadriel 1 0.13 0 7 0 7 0.06 Athila 1 0.13 0 14 0 14 0.12 Ty3-gypsy 743 100.00 5 275 6 861 1.30 12 136 100.00 总计 1 562 100.00 7 731 13 656 1.77 21 387 100.00 说明:a表示每个谱系的数量;c表示每个谱系在超家族中所占的比例;d表示结构完整的LTR反转录转座子(full-length LTR),包含 两端LTR序列和完整的编码结构域[44];e表示仅含有两端LTR序列,编码结构域有缺失的LTR反转录转座子(solo LTR)[44];f表 示每个谱系中full-length LTR和solo LTR在超家族中所占的比例 Table 2. Structure of LTR retrotransposon family of moso bamboo
根据LTR反转录转座子不同家族之间的进化关系和结构特征,Ty1-copia超家族和Ty3-gypsy超家族可以被分为多个不同的谱系[42-43]。根据Gypsy Database2.0[37]中植物典型的谱系序列特征,对毛竹LTR反转录转座子进行分类,将Ty1-copia超家族分为4个谱系,分别为Tork、Retrofit、Sire、Oryco;Ty3-gypsy超家族分为6个谱系,分别为Del、Reina、Crm、Tat、Galadriel、Athila。其中Tork包含236个家族,Reftrofit包含342个家族,Sire包含136个家族,Oryco包含105个家族,Del包含207个家族,Reina包含249个家族,Crm包含47个家族,Tat包含238个家族,Galadriel包含1个家族,Athila包含1个家族。在Ty1-copia超家族的4个谱系中,Sire的含量最高(达11.01%),紧随其后的是Tork(6.51%)。在Ty3-gypsy超家族的4个谱系中,Del的含量最高(达17.51%),紧随其后的是Tat(12.06%)(表1)。Tat和Del在植物中普遍存在并且是植物所特有的。ENV域在Sire中被识别,CHR域在Del和Reina中被识别(表1~2)。
-
在转座过程中,PBS是LTR反转录转座子反转录开始的重要位点,因为LTR反转录转座子开始反转录时tRNA会结合到RNA的PBS处,然后通过反转录酶合成cDNA[45]。不同超家族和谱系的LTR反转录转座子对PBS具有不同的偏好性。由表3所示:MetCAT24是转座子反转过程中使用频率最高的PBS位点,占4.05%,比其他的位点要高,其次是LysTTT和LysTTT10。表3中Ty1-copia和Ty3-gypsy超家族对PBS位点的偏好性呈相反趋势,MetCAT24是Ty1-copia超家族中使用最多的PBS位点,LysTTT是Ty3-gypsy超家族使用最多的PBS位点,但在Ty1-copia超家族中频率很低,仅有1个。LTR序列是LTR反转录转座子中特有的,它们位于LTR反转录转座子的5′端和3′端,是一对高度相似的序列,通常较长的LTR反转录转座子具有更长的LTR序列,结构也更加完整。所以把5′端LTR作为参照,对LTR序列长度进行统计,结果如图1显示。对LTR反转录转座子而言,LTR序列长度与其全长序列的长度成正比。
tRNA 数量/个 百分比/% Ty1-copia
使用比例/%Ty3-gypsy
使用比例/%MetCAT24 1 383 4.05 1.70 0.83 LysTTT 486 1.42 0.00 1.10 LysTTT10 285 0.83 0.31 0.03 LeuAAG21 131 0.38 0.00 0.15 LysTTT3 111 0.32 0.16 0.01 LeuTAG9 66 0.19 0.07 0.01 Table 3. Usage status of PBS in LTR retrotransposons
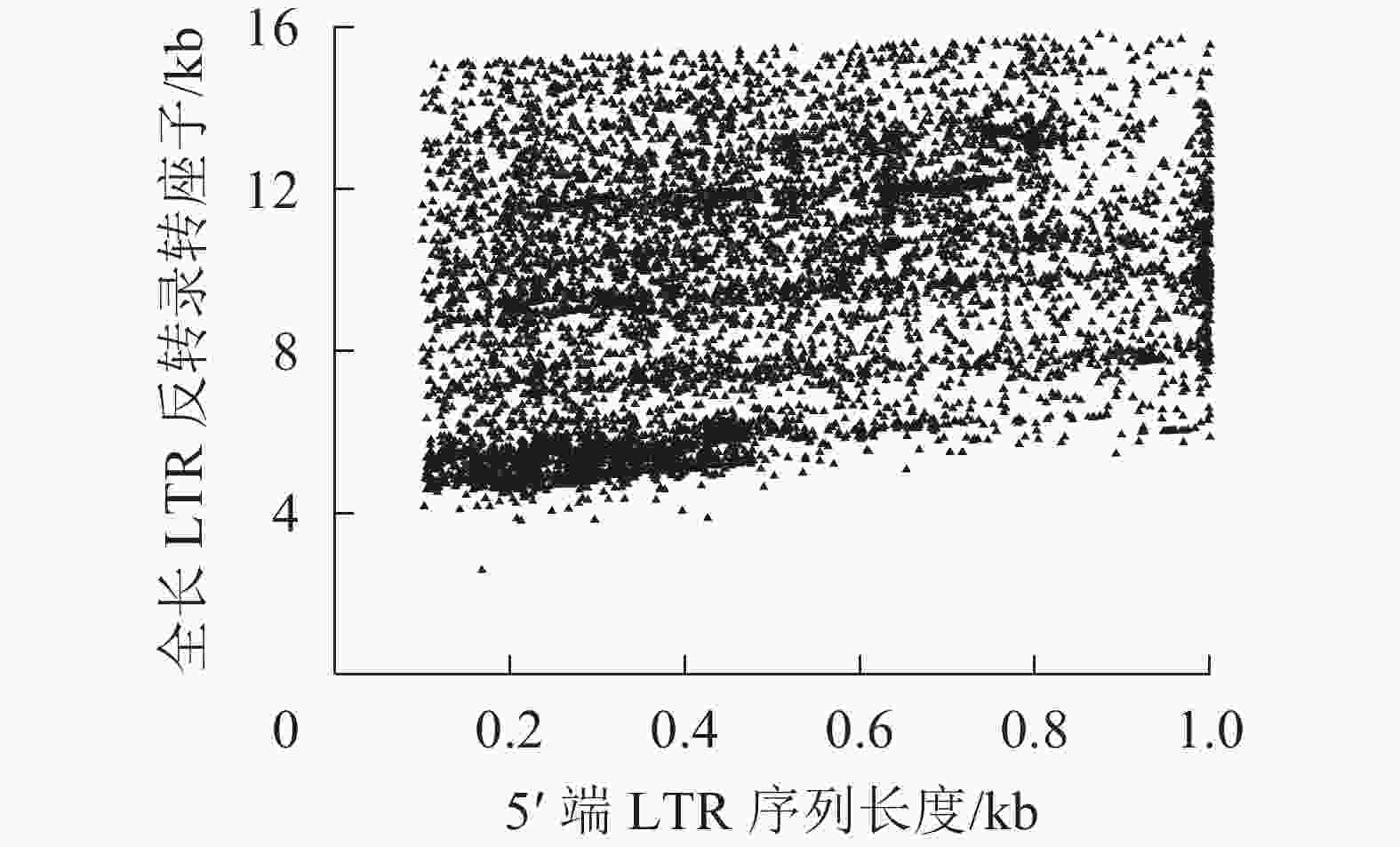
Figure 1. Correlation of length between LTR and LTR retrotransposons
-
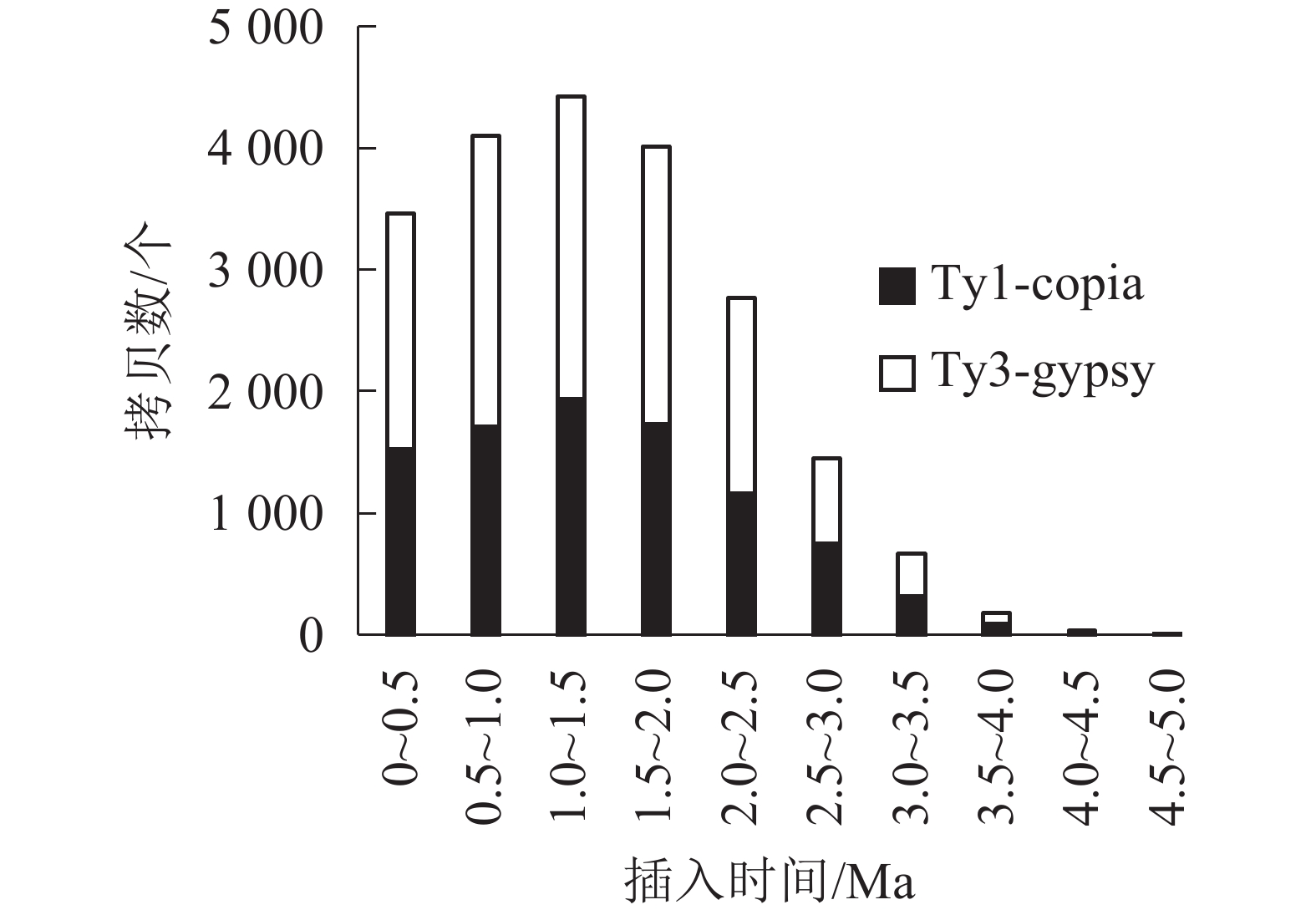
对21 387个含有TSD位点的毛竹LTR反转录转座子的插入时间进行统计,如图2和图3所示。毛竹LTR反转录转座子的插入时间集中于0~2.0 Ma,其中插入最旺盛的是在1.0~1.5 Ma,有4 426 个,占21.06%,插入较少的是在3.0 Ma之前,有891个,仅占2.61%,插入时间为0的有508个(占1.4%),说明这部分LTR反转录转座子可能还具有转座潜力。Del转座频率最高,有4 777个拷贝,占22.62%,且在0.5~1.5 Ma转座活动最为旺盛,其次为Sire和Tat。而Retrofit、Oryco、Reina、Crm转座频率都较低,Retrofit最低,仅有1 527个拷贝,占6.91%。以上数据说明毛竹基因组中LTR反转录转座子在0~2.0 Ma内大量复制增长,且还处于不断增长的状态,但增长趋势在减弱。
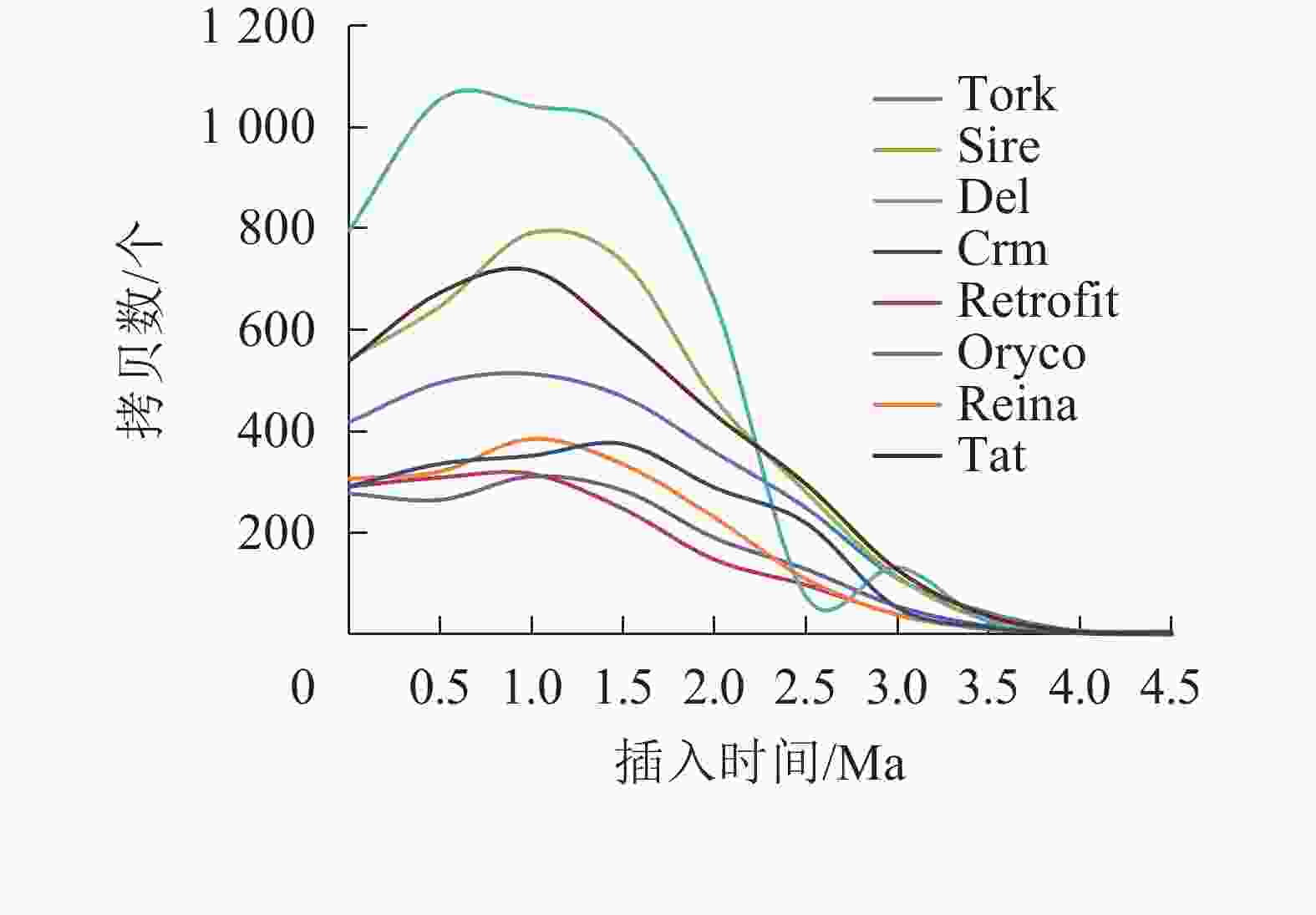
Figure 2. Insertion time distribution of different lineages of moso bamboo LTR retrotransposons

Figure 3. Insertion times of superfumily of moso bamboo LTR retrotransposons
2.1. 毛竹中LTR反转录转座子的鉴定和分类
2.2. 毛竹LTR反转录转座子的PBS偏好性及LTR序列长度分析
2.3. 毛竹LTR反转录转座子的插入时间分布

-
HU等[46]利用第1版毛竹基因组数据分析了LTR反转录转座子的分布结构和进化模式。但由于第1版毛竹基因组数据的碎片化严重,限制了对LTR反转录转座子的完整预测。毛竹第2版基因数据的覆盖范围、精准度都有所提高,完整性达95.2%[34],相比玉米(92.2%)[22]要高,与水稻(95.6%)[23]接近,所以本研究的结果准确性较高。然而,LTR反转录转座子是一种重复序列[47],软件无法准确逐条识别,产生假阴性。在不同的基因组中LTR反转录转座子所占的比例不同,如在酵母基因组中占3%[48],在玉米基因组中占70.1%[22],分布特点也有所差异,且基因组中还存在其他重复序列,所以也很容易产生假阳性。因此,利用较完整的毛竹第2版基因组,能在一定程度上避免误差。
在本研究中,通过对第2版毛竹基因组的注释共得到1 014 565条LTR反转录转座子(表1),占基因组的54.7%,较对第1版毛竹基因组注释的结果(1 954 616,39.83%,不包括未知LTR)数量有所下降,比例有所上升。对7 731条结构完整的LTR反转录转座子进行家族分类,共划分为1 562个家族,较第1版(959个)也有所上升。通过对LTR反转录转座子的结构完整性判断,solo-LTR较完整LTR反转录转座子比例更高(S/F为1.77),说明可能毛竹LTR反转录转座子不平衡重组和非法重组的活动频率较高,这可能是由于毛竹基因组在不断进化的过程中对转座子产生抑制,碎片化严重。但在第1版毛竹基因组中完整的LTR反转录转座子的占比更高(S/F为0.28),这是由于第1版基因组不完整所以信息显示不全面。总的来说,第2版毛竹基因组在转座子的注释、鉴定、家族和超家族的分类等方面都相比第1版毛竹基因组更加准确,但是依然存在数万条短的组装碎片,因此本研究对毛竹LTR反转录转座子的注释仍是保守值,随着毛竹基因组的更加完善会有更多的LTR反转录转座子被发现。
-
根据Gypsy Database 2.0网站中植物典型的谱系序列特征,毛竹LTR反转录转座子共分为Sire、Oryco、Retrofit、Tork、Crm、Del、Reina、Tatol、Galadriel、Athila等10个谱系。其中Retrofit、Reina和Tat是数量最多的3个谱系。Galadriel和Athila在植物界中虽然广泛存在[15],但在毛竹基因组发现较少。一方面可能是Athila的大小通常为8.5~12.0 kb,并且具有相对较长的LTR序列(1.5~2.5 kb),很难被LTRhavest程序识别[35];另一方面,毛竹基因组的不完整和碎片化,可能会遗漏Galadriel和Athila。
将毛竹LTR反转录转座子的谱系与水稻[47]、拟南芥Arabidopsis thaliana[49]进行比较,发现毛竹的10个谱系包含了双子叶植物和单子叶植物共同的进化特征,这是它们分裂后分化的结果[50],其中Tork是例外,它在毛竹、水稻、拟南芥中并无明显差异,这表明Tork相对其他谱系更加保守。并且毛竹与水稻亚科在LTR反转录转座子都为多拷贝和多进化谱系并存,而拟南芥的谱系数则相对较少[49],这可能是由于不同进化速率导致的结果。
在不同谱系之间,LTR反转录转座子增殖差异较大。在毛竹Ty1-copia超家族中,Retrofit的LTR反转录转座子家族数量最多(342个),但拷贝数仅占15.78%,相比之下,Sire只有136个家族,拷贝数却达11.1%,占比最高。这种情况在Ty3-gypsy超家族更为显著,Reina包含249个家族,拷贝数却仅占2.06%,Crm只有47个家族,拷贝数所占比例比Reina还高一些。各谱系中LTR反转录转座子的数量反映了它们最近的扩增情况,而各谱系中家族的数量则代表了历史上不同的分化情况。因此,不同谱系的LTR反转录转座子在进化过程中具有不同的分化和扩增活性。
-
LTR反转录转座子两端的LTR序列是相同的,但在不断转座过程中,LTR序列会发生突变并分化,根据剪辑替换速率,可以得出LTR反转录转座子的插入时间[31]。毛竹LTR反转录转座子的插入时间在0~1.5 Ma呈直线式递增,但在大于1.5 Ma呈指数衰减,总体上呈现抛物线形式,与小麦Triticum aestivum [51]、桑树Morus notabilis[52]类似,这说明毛竹LTR反转录转座子的插入时间主要集中于0~2.0 Ma,在1.5 Ma处于转座活动爆发期,但其增长趋势处于回缩的状态。
毛竹LTR反转录转座子在大于5.0 Ma区域缺失,可能原因:第一,5.0 Ma之前的老LTR反转录转座子与年轻的LTR反转录转座子发生重组,所以无法识别[42];第二,根据PENG等[33]的研究,在7.0~12.0 Ma中,毛竹基因组发生了四倍体事件,之后又不断进化为二倍体,在这个过程中毛竹基因组经历了较大的选择压力,所以5.0 Ma之前的毛竹LTR反转录转座子在基因组中被删除或严重破坏,无法通过结构预测和同源性比对来鉴定。
 DownLoad:
DownLoad: