





-
‘怀玉山’高山马铃薯Solanum tuberosum var. cormosus ‘Huaiyushan’,又名麻籽洋芋,茄科Solanaceae茄属Solanum 1年生草本植物,主要种植区域为江西省玉山县怀玉乡[1]。‘怀玉山’高山马铃薯食用、药用皆优,获批为国家地理标志农产品[2−3]。已有研究表明:‘怀玉山’高山马铃薯与云南德宏和曲靖以及湖北恩施的高山马铃薯种质存在差异[4]。但‘怀玉山’高山马铃薯的进化来源尚无相关研究报道。
叶绿体是高等植物细胞内一种重要的与光合作用和物质代谢相关的细胞器,叶绿体基因组是一套具有母系遗传特征的独立基因组,是高等植物细胞质基因组的组成成分之一[5]。与核基因组相比,叶绿体基因组全长序列短、易测序获得、基因直系同源、基因结构稳定、保守性较高、进化速率适中,目前已经广泛应用于植物系统发育分析、物种分类鉴定及分子标记开发等研究中,在物种起源、进化、演变及比较基因组学等研究领域发挥着越来越大的作用[6]。密码子是核酸和蛋白质之间遗传信息传递的桥梁[7],mRNA上的遗传信息以tRNA三重密码子传递。氨基酸一般对应≥1的密码子[8],这些密码子称为同义密码子[9]。在自然选择或突变偏好的情况下,基因倾向于使用≥1的同义密码子,即同义密码子使用偏好性[10−12]。目前,关于茄属的叶绿体基因组研究已有报道[13−19],而针对‘怀玉山’高山马铃薯的研究大多集中在基因克隆[20]、转录组分析[3]、遗传多样性[4]、脱毒快繁[2]、DNA甲基化敏感扩增多态性(MSAP)分析[21]等方面,对‘怀玉山’高山马铃薯叶绿体全基因组及其密码子使用偏好性方面的研究还未见系统报道。本研究通过对‘怀玉山’高山马铃薯叶绿体基因组进行测序和组装,分析基于叶绿体基因组的‘怀玉山’高山马铃薯系统进化、结构解析和密码子偏好性等,为‘怀玉山’高山马铃薯叶绿体基因组研究和应用提供科学依据,也为进一步研究‘怀玉山’高山马铃薯遗传背景、种质资源保护与开发利用奠定基础。
-
由上饶市薯芋类作物种质保存与利用重点实验室提供的‘怀玉山’高山马铃薯试管苗。
-
选取‘怀玉山’高山马铃薯(MLS)试管苗叶片组织,利用植物基因组DNA提取试剂盒(北京天根生化科技有限公司)提取‘怀玉山’高山马铃薯试管苗DNA,质量分数为1%琼脂糖凝胶电泳检测DNA的完整性,NanoDrop 2000 分光光度计(Thermo Scientific公司)检测 DNA 浓度和纯度,用超声波将DNA片段化,然后对片段化的DNA进行片段纯化、末端修复、3′端加A、连接测序接头,再用琼脂糖凝胶电泳进行片段大小选择,进行聚合酶链式反应(PCR)扩增形成测序文库。建好的文库先进行文库质检,质检合格的文库用BGISEQ-500平台进行测序。
-
通过SOAPnuk 1.3.0对raw data (测序下机的原始数据)进行数据过滤,去除其中的接头序列及低质量reads (高通量测序中一个反应获得的测序序列),获得高质量的clean data (对原始数据进行过滤后并剔除了低质量数据的剩余数据)。采用Noveplastys软件组装叶绿体基因组核心模块,以起始组装序列为起点开始组装叶绿体contigs (很多reads根据序列拼接在一起拼出的片段),如果contigs未环化,则利用CAP 3软件连接多个contigs为完整叶绿体基因组,并手动调整环状叶绿体基因组起始位置。使用GeSeq、tRNAscan-SE对叶绿体基因组进行注释,再经过手工校正后得到最终的基因注释结果。将注释完成的‘怀玉山’高山马铃薯叶绿体基因组序列提交至美国国家生物信息中心(NCBI),获得登录号:OP589401。使用OGDRAW绘制叶绿体基因组图谱。
-
通过JSHYCloud在线工具集分析并统计叶绿体基因组、大单拷贝区(LSC)、小单拷贝区(SSC)和反向重复区(IR)的鸟嘌呤和胞嘧啶所占的比例(GC比例);使用MISA软件进行简单重复序列(SSR)分析,单核苷酸、二核苷酸、三核苷酸、四核苷酸、五核苷酸、六核苷酸的最小重复值分别设置为10、6、5、5、5、5;利用REPuter软件进行长重复序列(longrepeat)分析,查找正向重复(F)、反向重复(R)、互补重复(C)、回文重复(P)等4种重复类型;通过Pasteur Galaxy 在线工具集中的CodonW模块分析密码子使用情况,设置输出结果为有效密码子数(ENC)和相对同义密码子使用频率(RSCU),其他参数设为默认值。将‘怀玉山’高山马铃薯叶绿体基因组序列上传至美国国家生物技术信息中心(NCBI) 进行BLASTn比对,选择highly similar sequence (megablast)比较相似性在95 %以上的序列,检索获得‘怀玉山’高山马铃薯的近缘种。利用Gview、VISTA tools、IRscope和DNADnaSP 6.0软件绘制‘怀玉山’高山马铃薯及其10个近缘种(S. cochoae NC_062512、多毛番茄S. habrochaites NC_026879、潘那利番茄S. pennellii NC_035742、S. bukasovii MT120867、S. boliviense NC_062870、S. trisectum NC_062469、S. salamancae NC_062480、S. clivorum NC_062513、S. mortonii NC_062426、S. insanum MW384851)的变异圈图、mVIST结构变异图、IR结构变异图,计算‘怀玉山’高山马铃薯及其10个近缘种的基因组核酸多样性(Pi),参数设置100 bp滑窗,25 bp的步长,并进行中性绘图分析(GC3-GC12分析)、ENC-plot分析、PR2-bias-plot分析和最优密码子分析;对‘怀玉山’高山马铃薯叶绿体基因的ENC进行排序,分别选取两端基因各5个,构建高表达基因库(ENC小)和低表达基因库(ENC大),并计算两者的RSCU差值(ΔRSCU)。筛选ΔRSCU≥0.08的高表达密码子,且将RSCU>1.00的高频率密码子定义为‘怀玉山’高山马铃薯叶绿体基因组的最优密码子;最后利用mafft 7.0和fasttree 2.1.10软件分别对‘怀玉山’高山马铃薯和18个近缘种以及烟草属Nicotiana 2个外类群物种进行序列比对和构建进化树。
-
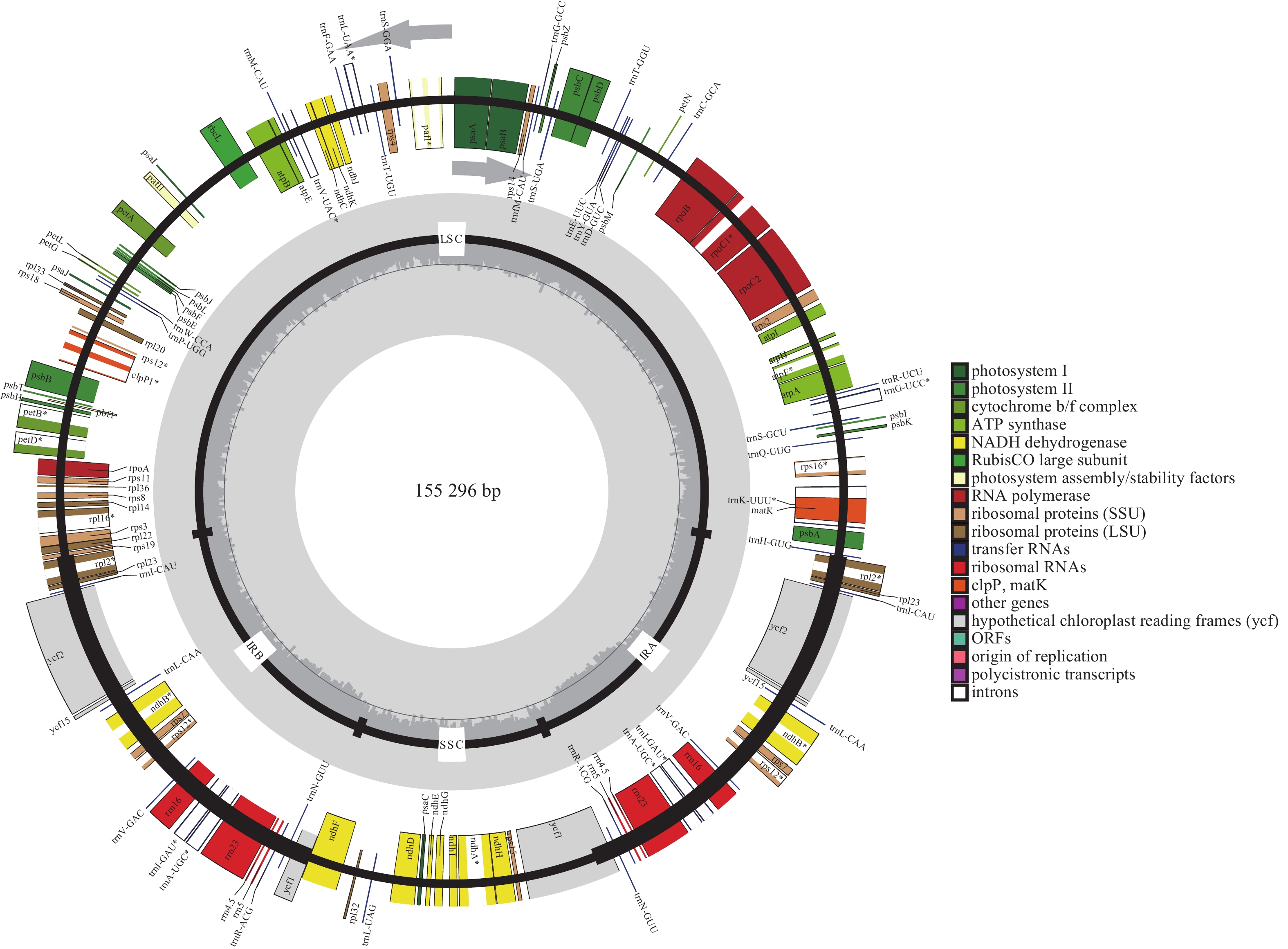
经过测序组装的完整的叶绿体基因组长度为155 296 bp,图1显示:‘怀玉山’高山马铃薯叶绿体基因组呈典型的四分体结构,包含1个LSC、1个SSC和2个将LSC与SSC分隔开的IR (IRa和IRb)。基因组的总GC比例为37.88%,A、T、C、G比例分别为30.65%、31.47%、19.24%、18.65%。LSC、SSC和IR的长度分别为85 737、18 373、25 593 bp。LSC的GC比例为36.01%,A、T、C、G比例分别为31.29%、32.70%、18.40%、17.61%;SSC的GC比例为32.09%,A、T、C、G比例分别为33.78%、34.14%、16.69%、15.40%;IRb的GC比例为43.10%,A、T、C、G比例分别为28.57%、28.33%、20.72%、22.39%;IRa的GC比例为43.10%,A、T、C、G比例分别为28.33%、28.57%、22.39%、20.72%。表明‘怀玉山’高山马铃薯IR的GC比例最大,LSC次之,SSC最少;叶绿体基因组总GC比例显著低于AT比例;叶绿体基因组各碱基比例从大到小依次为T、A、C、G。
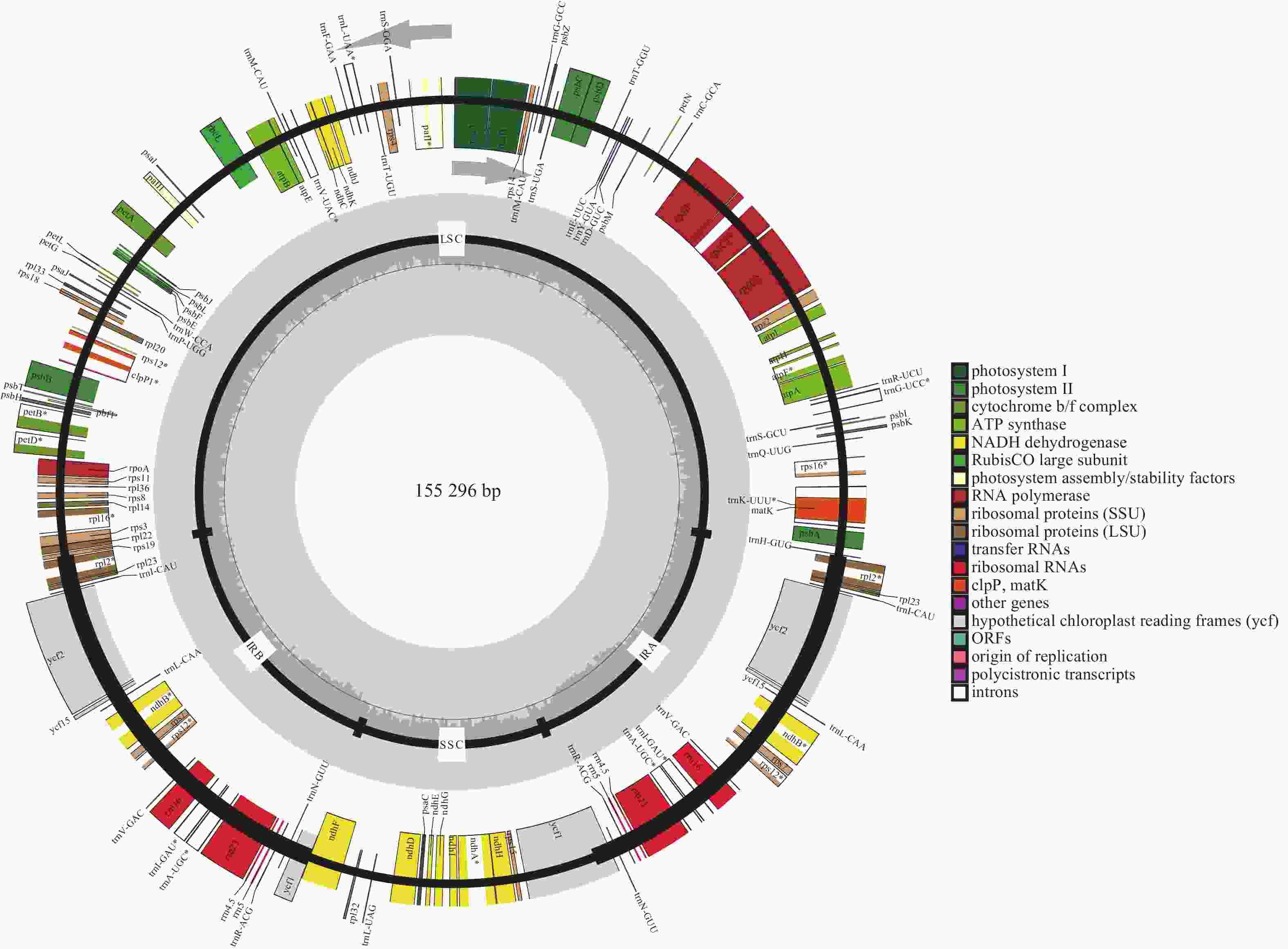
Figure 1. Chloroplast genome map of S. tuberosum var. cormosus ‘Huaiyushan’
-
叶绿体基因组共注释到光合作用基因、自我复制基因、其他基因和未知功能基因4类,包括87个编码区(CDS)基因、37个tRNA基因、8个rRNA 基因、1个假基因,共133个基因。对有多个外显子的叶绿体基因进行结构分析,由2个外显子构成的基因有21个,包括13个CDS基因和8个tRNA基因;由 3个外显子构成的基因有4个,为clpP1、ycf3、rps12 (2个)基因。LSC的基因数量最多(81个),其中CDS基因59个、tRNA基因22个;SSC的基因数量为11个,其中CDS基因10个、tRNA基因1个;IR的基因数量为17个,其中CDS基因6个、rRNA基因4个、tRNA基因7个;SSC与IRb边界(JSB)的基因数量为2个(ndhF和ycf1);LSC与IRb边界(JLB)的基因数量为1个(rps19);SSC与IRa边界(JSA)的基因数量为2个(ycf1);LSC与IRa边界(JLA)的基因数量为0。rps12有2个拷贝,每个拷贝具有3个外显子,且2个拷贝共享第1个外显子,第1个外显子位于LSC,另外2个外显子位于IR (表1)。
基因功能 基因类型 基因名 基因数
量/个光合作用 光系统Ⅰ psaA、psaB、psaC、psaI、psaJ 5 光系统Ⅱ psaJ、psbA、psbB、psbC、psbD、psbE、psbF、psbH*、psbI、psbK、psbL、psbM、
psbT、psbZ15 NADH 脱氢 ndhA、ndhB*、ndhC、ndhD、ndhE、ndhF、ndhG、ndhH、ndhI、ndhJ、ndhK 12 细胞色素 b/f 复合体 petA、petB、petD、petG、petL、petN 6 ATP 合成酶 atpA、atpB、atpE、atpF、atpH、atpI 6 自我复制 核糖体大亚基蛋白质 rpl14、rpl16、rpl2*、rpl20、rpl22、rpl23*、rpl32、rpl33、rpl36 11 核糖体小亚基蛋白质 rps11、rps12*、rps14、rps15、rps16、rps18、rps19、
rps2、rps3、rps4、rps7*、rps814 核糖体大亚基 rbcL 1 RNA 聚合酶 rpoA、rpoB、rpoC1、rpoC2 4 核糖体RNA rrn16*、rrn23*、rrn4.5*、rrn5* 8 转运RNA trnA-UGC*、trnC-GCA、trnD-GUC、trnE-UUC、trnF-GAA、trnG-GCC、trnG-UCC、
trnH-GUG、trnI-CAU*、trnI-GAU*、trnK-UUU、trnL-CAA*、trnL-UAA、trnL-UAG、
trnM-CAU、trnN-GUU*、trnP-UGG、trnQ-UUG、trnR-ACG*、trnR-UCU、
trnS-GCU、trnS-GGA、trnS-UGA、trnT-GGU、trnT-UGU、trnV-GAC*、trnV-UAC、
trnW-CCA、trnY-GUA、trnfM-CAU37 其他基因 成熟酶 matK 1 蛋白酶 clpP1 1 囊膜蛋白 cemA 1 乙酰辅酶 A 羧化酶 accD 1 c-型细胞色素合成基因 ccsA 1 翻译起始因子 infA 1 未知功能基因 保守假设叶绿体阅读框架 ycf1*、ycf15*、ycf2*、ycf3、ycf4 8 说明:*表示该基因的数量有2个。 Table 1. Chloroplast gene functional classification of S. tuberosum var. cormosus ‘Huaiyushan’
-
叶绿体基因组中共检测到38个SSR位点,其中,单碱基重复有36个,双碱基重复有2个。其中,重复单元为A/T,重复频率为10的SSR位点数量最多(18个),重复频率为11的SSR位点数量次之(11个);重复单元为AT/AT、重复频率为6的SSR位点数量为2个。
-
叶绿体基因组共鉴定到32个长重复序列,包括16个正向重复(15个30~39 bp,1个40~49 bp),16个回文重复 (13个30~39 bp,2个40~49 bp,1个50~59 bp),无反向重复和互补重复。
-
‘怀玉山’高山马铃薯及其10个近缘种叶绿体基因组结构从LSC中间呈线性展开,均由1个LSC、1个SSC和2个IR (IRa和IRb) 4部分组成。‘怀玉山’高山马铃薯及其10个近缘种rpl22、rps19、rpl2、ycf1、ndhF、trnH和psbA位置基本一致,但收缩和扩张的长度存在一些差异(图2)。
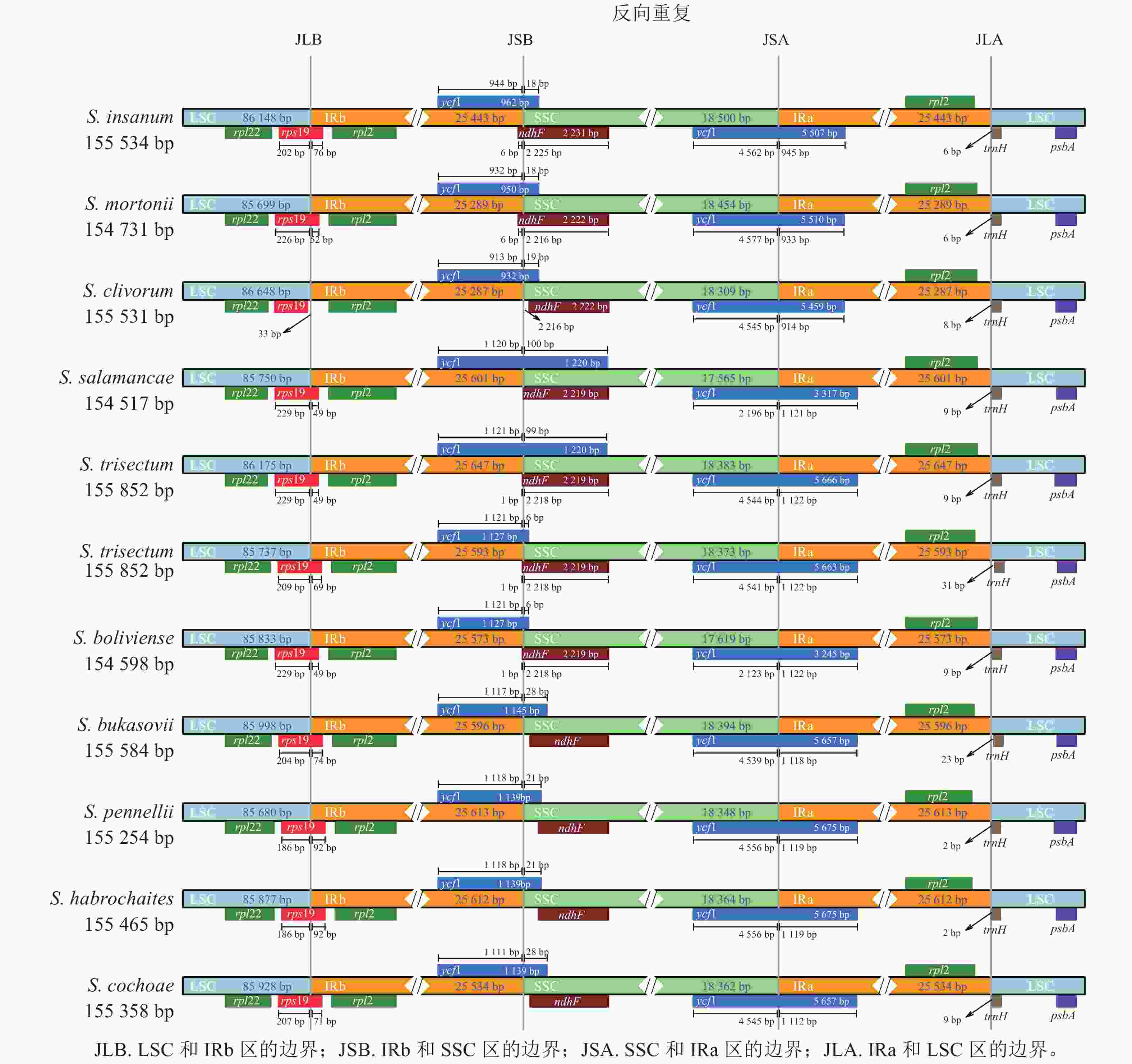
Figure 2. Comparison on the boundary locations of large single copy region, small single copy region and inverted repeat region in chloroplast genomes of S. tuberosum var. cormosus ‘Huaiyushan’ and its 10 related species
-
‘怀玉山’高山马铃薯及其10个近缘种叶绿体基因组核苷酸多样性的变化范围为0~0.13927,高变区主要分布在LSC和SSC。LSC的trnL-UAA-trnF-GAA、cemA、rps12-exon1-clpP1、clpP1基因变异率最高;SSC的rpl32-trnL-UAG、ycf1基因变异率最高。
-
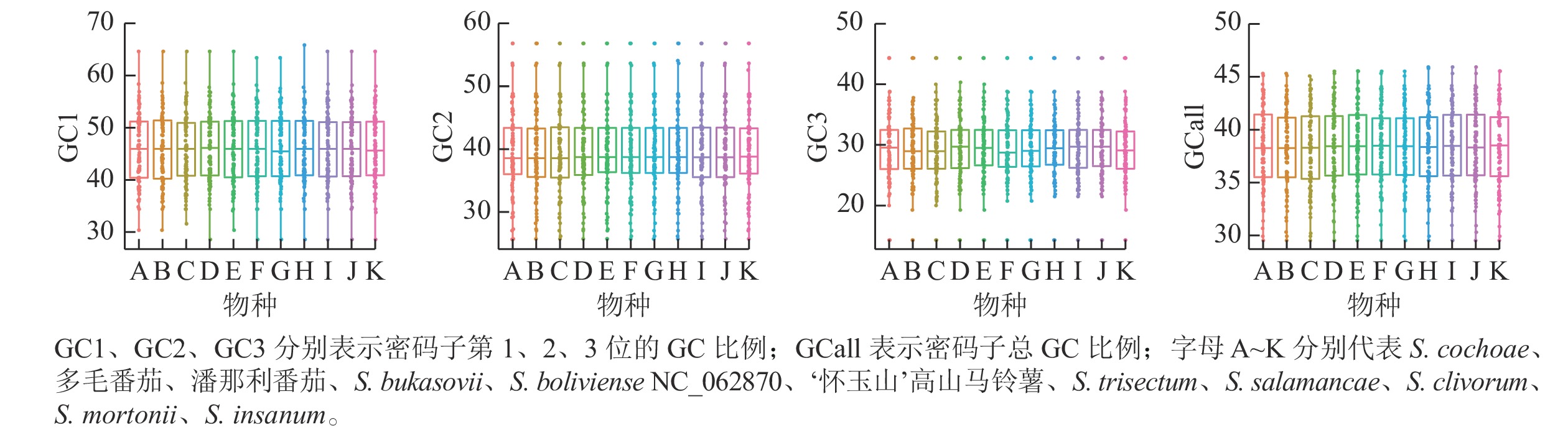
叶绿体基因组87个CDS基因密码子3个位置GC比例的平均值为38.38%,GC1、GC2、GC3分别为45.98%、39.55%、29.60%,这说明GC在密码子3个位点上的分布存在显著差异,只有GC2与平均GC大致接近(图3)。ENC是密码子偏性分析的重要指标,通常将35作为区分值来评估密码子偏倚的强度。叶绿体基因组87个CDS 基因的平均ENC为47.29,ENC>45的基因有60个,ENC>35的基因有83个,有4个基因的ENC<35,这表明叶绿体基因组的密码子偏性较弱。通过SPSS 20.0进行相关性分析,结果表明:密码子总GC比例(GCall)与GC1、GC2在0.01水平上均存在极显著的正相关,GCall与GC3在0.05水平上显著相关;GC1与GC2在0.05水平上存在显著正相关,但两者均与GC3不相关。这表明叶绿体基因组密码子前2位的碱基组成相似,而与第3位不相似。ENC与GC1、GC2、GC3均不相关,说明密码子上第1位、第2位和第3位的碱基组成对ENC没有显著影响。叶绿体基因组 87个CDS基因序列共有31个RSCU>1的密码子。在这31个密码子中,除AUG、UUG外,其余都以A、U结尾,表明A、U碱基在密码子最后位点上出现的频率最高。‘怀玉山’高山马铃薯叶绿体基因组密码子偏好以A、U结尾(表2)。
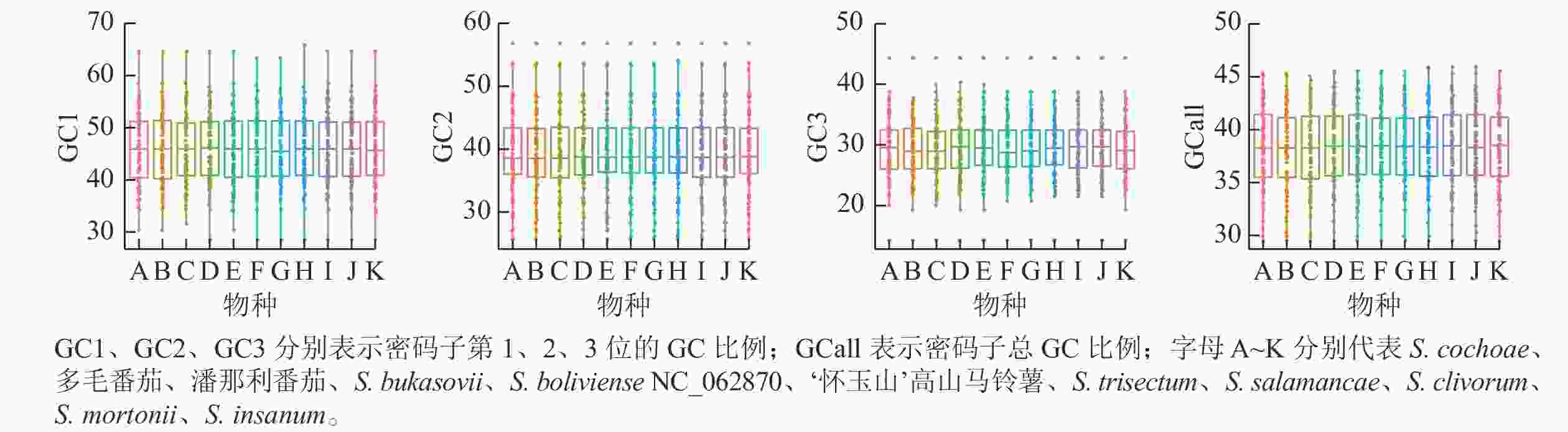
Figure 3. Composition analysis of chloroplast genome codons of S. tuberosum var. cormosus ‘Huaiyushan’ and its 10 related species
密码子 氨基酸 相对同义密码子
使用频率数量/个 密码子 氨基酸 相对同义密码子
使用频率数量/个 密码子 氨基酸 相对同义密码子
使用频率数量/个 GCA Ala 1.134 37 401 GGG Gly 0.731 07 333 CCU Pro 1.521 13 432 GCC Ala 0.718 53 254 GGU Gly 1.242 59 566 AGC Ser 0.342 61 119 GCG Ala 0.390 38 138 CAC His 0.479 87 149 AGU Ser 1.191 94 414 GCU Ala 1.756 72 621 CAU His 1.520 13 472 UCA Ser 1.197 70 416 AGA Arg 1.829 81 491 AUA Ile 0.909 09 680 UCC Ser 0.955 85 332 AGG Arg 0.633 54 170 AUC Ile 0.609 63 456 UCG Ser 0.595 97 207 CGA Arg 1.453 42 390 AUU Ile 1.481 28 1 108 UCU Ser 1.715 93 596 CGC Arg 0.368 94 99 CUA Leu 0.821 14 391 UAA Ter 1.655 17 48 CGG Arg 0.424 85 114 CUC Leu 0.434 72 207 UAG Ter 0.758 62 22 CGU Arg 1.289 44 346 CUG Leu 0.403 22 192 UGA Ter 0.586 21 17 AAC Asn 0.485 74 315 CUU Leu 1.297 86 618 ACA Thr 1.221 17 421 AAU Asn 1.514 26 982 UUA Leu 1.812 39 863 ACC Thr 0.771 57 266 GAC Asp 0.408 80 223 UUG Leu 1.230 66 586 ACG Thr 0.446 70 154 GAU Asp 1.591 20 868 AAA Lys 1.462 87 1 054 ACU Thr 1.560 55 538 UGC Cys 0.556 29 84 AAG Lys 0.537 13 387 UGG Trp 1.000 00 490 UGU Cys 1.443 71 218 AUG Met 1.987 34 628 UAC Tyr 0.397 12 193 CAA Gln 1.491 10 712 GUG Met 0.012 66 4 UAU Tyr 1.602 88 779 CAG Gln 0.508 90 243 UUC Phe 0.722 19 542 GUA Val 1.502 75 547 GAA Glu 1.477 52 1 035 UUU Phe 1.277 81 959 GUC Val 0.52473 191 GAG Glu 0.522 48 366 CCA Pro 1.193 66 339 GUG Val 0.524 73 191 GGA Gly 1.571 90 716 CCC Pro 0.742 96 211 GUU Val 1.447 80 527 GGC Gly 0.454 45 207 CCG Pro 0.542 25 154 Table 2. Relative synonymous codon usage (RSCU) of chloroplast genome of S. tuberosum var. cormosus ‘Huaiyushan’
-
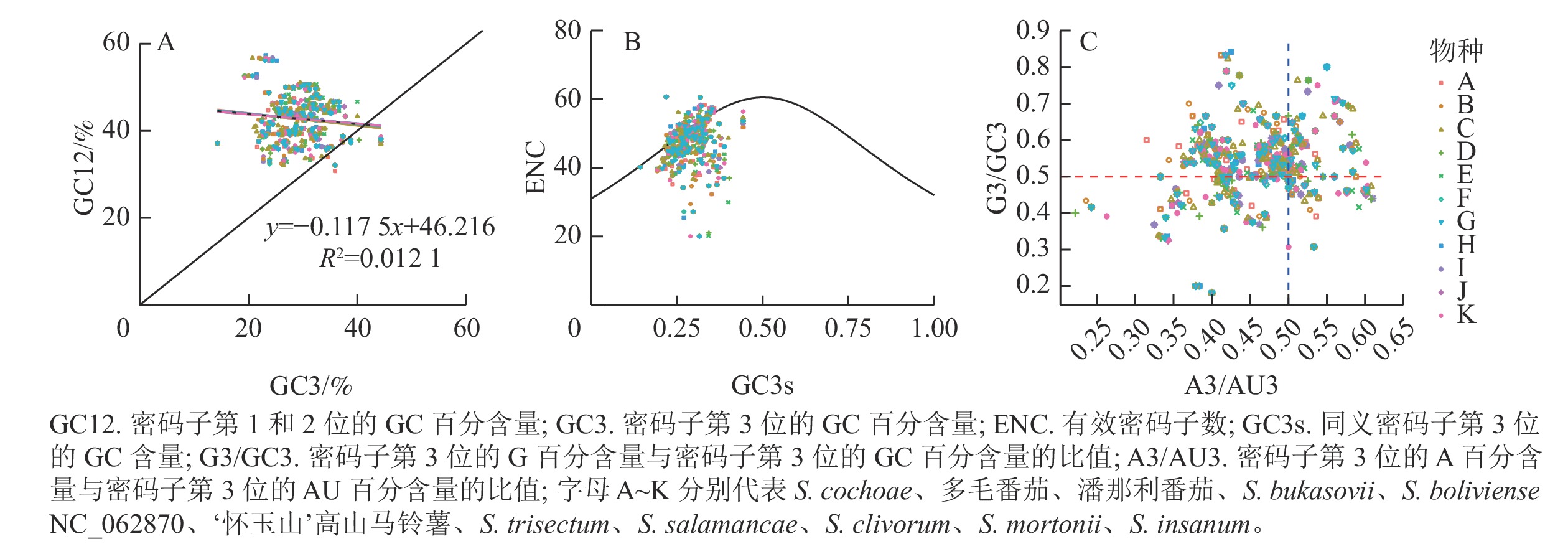
分析发现:‘怀玉山’高山马铃薯及其10个近缘种植物叶绿体基因的GC3比例分布为0.142 9~0.443 2,GC12比例分布为0.285 7~0.658 5,两者大多沿对角线上方分布。两者的相关系数(r)为0.110 1 (R2=0.012 1),相关不显著(P>0.05),回归斜率为0.117 5,说明GC12与GC3不相关(图4A)。表明‘怀玉山’高山马铃薯叶绿体基因组密码子使用偏性很大程度上受自然选择的影响,而受突变压力的影响小。

Figure 4. GC3-GC12 analysis (A), ENC-plot analysis (B) and PR2-plot analysis (C) of chloroplast genome codons of S. tuberosum var. cormosus ‘Huaiyushan’ and its 10 related species
-
分析表明:分布在期望曲线上或曲线附近的基因较少,分布在期望曲线下方且远离曲线的基因较多,说明大部分基因的实际ENC (ENCobs)与理论ENC (ENCexp)存在差异。为了解实际ENC和理论ENC的差异度,计算了‘怀玉山’高山马铃薯ENC比值频数,即(ENCexp-ENCobs)/ENCexp。结果表明:‘怀玉山’高山马铃薯叶绿体基因组基因中,有16.47%(14个)的基因分布在0~0.1区间,分布于期望曲线上或曲线附近,即ENCobs接近于ENCexp值,有83.53%的基因分布在0~0.1区间外,远离期望曲线分布,即ENCexp和ENCobs相差较大,表明自然选择是影响‘怀玉山’高山马铃薯叶绿体基因组密码子使用偏性的主要因素,而突变压力的作用较小(图4B)。
-
分析表明: A3/AU3轴、G3/GC3轴均以0.5为界限,发现4个平面内基因分布不均衡。从G3/GC3轴看,多数基因位于上方(>0.5),少数基因位于下方(<0.5);从A3/AU3轴看,多数基因位于左侧(<0.5),少数基因位于右侧(>0.5)。这表明4种碱基在同义密码子第3位上存在C>G、T>A现象(图4C)。当密码子使用存在偏性完全受突变压力影响时,C和G以及A和T同义密码子在第3位上的分布应相等。因此,‘怀玉山’高山马铃薯叶绿体基因组密码子使用偏性主要受自然选择等因素影响。
-
RSCU分析可知:同时满足RSCU>1和ΔRSCU≥0.08的密码子共10个,即CGU、AAA、CUU、GUU、GGA、GUA、GGU、UCA、GCU、CCU,这些密码子都以A、U结尾,被确定为‘怀玉山’高山马铃薯叶绿体基因组的最优密码子(表3)。
密码子 氨基酸 相对同义密
码子使用率密码子高表
达相对概率密码子低表
达相对概率ΔRSCU 密码子 氨基酸 相对同义密
码子使用率密码子高表
达相对概率密码子低表
达相对概率ΔRSCU CGU* Arg 1.289 44 0.631 58 1.428 57 0.796 99 UUU Phe 1.277 81 1.315 79 1.200 00 −0.115 79 AAA* Lys 1.462 87 1.000 00 1.750 00 0.750 00 UUG Leu 1.230 66 0.750 00 0.600 00 −0.150 00 CUU* Leu 1.297 86 1.022 73 1.500 00 0.477 27 AUU Ile 1.481 28 1.428 57 1.263 16 −0.165 41 GUU* Val 1.447 80 0.800 00 1.250 00 0.450 00 CAU His 1.520 13 1.333 33 1.000 00 −0.333 33 GGA* Gly 1.571 90 1.302 33 1.750 00 0.447 67 GAA Glu 1.477 52 1.469 39 1.000 00 −0.469 39 GUA* Val 1.502 75 1.400 00 1.750 00 0.350 00 ACA Thr 1.221 17 1.000 00 0.444 44 −0.555 56 GGU* Gly 1.242 59 1.302 33 1.500 00 0.197 67 CCA Pro 1.193 66 1.142 86 0.571 43 −0.571 43 UCA* Ser 1.197 70 1.000 00 1.153 85 0.153 85 GAU Asp 1.591 20 1.600 00 1.000 00 −0.600 00 GCU* Ala 1.756 72 2.105 26 2.250 00 0.144 74 AAU Asn 1.514 26 1.600 00 1.000 00 −0.600 00 CCU* Pro 1.521 13 1.571 43 1.714 29 0.142 86 CAA Gln 1.491 10 1.615 38 1.000 00 −0.615 38 AUG Met 1.987 34 1.000 00 1.000 00 0.000 00 AGU Ser 1.191 94 1.444 44 0.769 23 −0.675 21 UAA Ter 1.655 17 1.200 00 1.200 00 0.000 00 CGA Arg 1.453 42 1.421 05 0.714 29 −0.706 76 UGU Cys 1.443 71 1.000 00 1.000 00 0.000 00 UUA Leu 1.812 39 1.704 55 0.900 00 −0.804 55 ACU Thr 1.560 55 1.600 00 1.555 56 −0.044 44 UCU Ser 1.715 93 1.888 89 0.769 23 −1.119 66 GCA Ala 1.134 37 0.421 05 0.375 00 −0.046 05 AGA Arg 1.829 81 2.368 42 1.071 43 −1.296 99 UAU Tyr 1.602 88 1.548 39 1.500 00 −0.048 39 说明:标注*的密码子为最优密码子。 Table 3. Optimal codon screening of chloroplast genome of S. tuberosum var. cormosus ‘Huaiyushan’
-
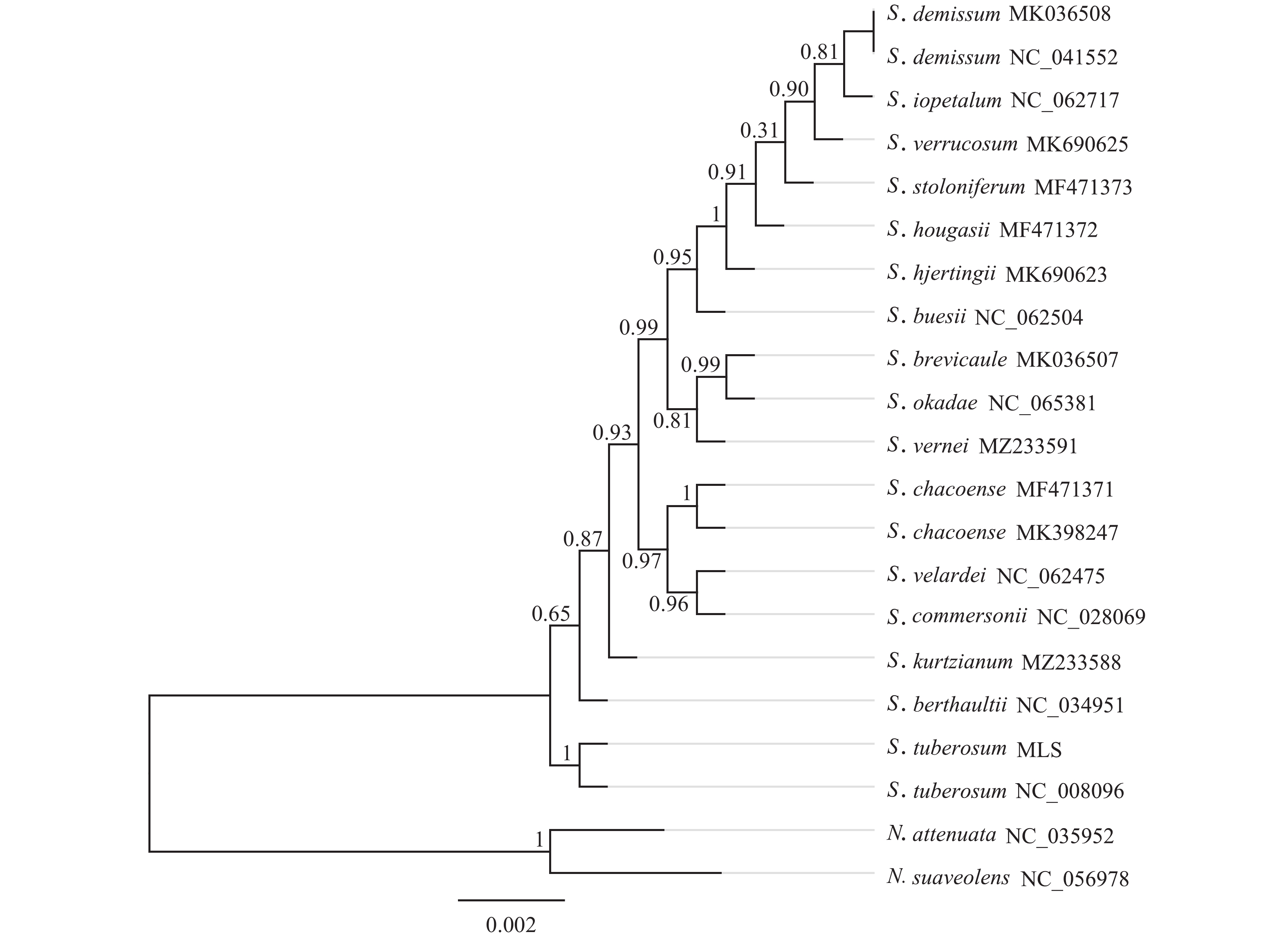
基于‘怀玉山’高山马铃薯和18个近缘种以及烟草属2个外类群物种叶绿体基因组构建的系统发育树分析可知:茄属聚为一大类,烟草属聚为另一大类。在茄属中,MLS与S. tuberosum NC_008096 (‘Ddeiree’)聚为一小分支。说明‘怀玉山’高山马铃薯与S. tuberosum ‘Ddeiree’亲缘关系较近,两者同源(图5)。
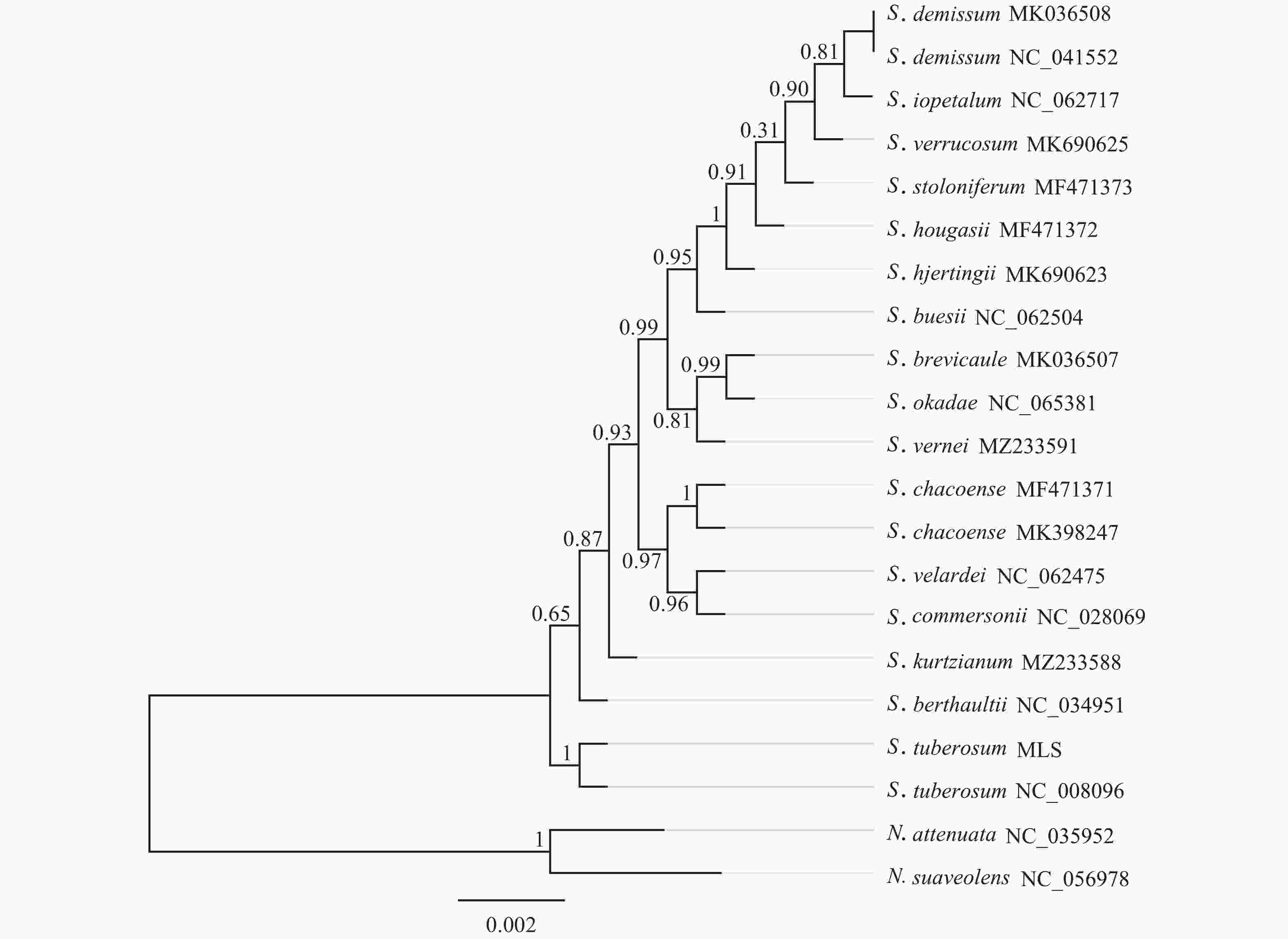
Figure 5. Phylogenetic tree of S. tuberosum var. cormosus ‘Huaiyushan’ and its 18 related species based on chloroplast genome
-
叶绿体基因组结构保守、独立母系遗传,是被子植物基因组的重要组成部分,广泛用于被子植物的生长发育、类群分析和进化分析[22]。被子植物叶绿体基因组大小一般为120~180 kb,IR大小一般为20~30 kb [23−24]。在本研究中,‘怀玉山’高山马铃薯叶绿体基因组长度和IR长度分别为155 296和25 593 bp,与S. tuberosum ‘Shepody’[16]叶绿体基因组长度和IR长度一致,与其他马铃薯品种[13−15, 17−18]相比,叶绿体基因组长度和IR长度不超过500 bp,说明马铃薯各个品种的叶绿体基因组较为保守。
叶绿体的SSR不仅与核基因组SSR一样,具有高多态性、多等位性、共显性[25],也具有单亲遗传模式,结构简单、相对保守[26],因此,叶绿体的SSR有较好的种间、种内遗传变异区分能力,已成为区分物种的重要分子标记而被广泛应用[27]。关惜今等[13]研究表明:S. fernandezianum与其野生近缘种(S. phureja、S. palustre、S. etuberosum)叶绿体基因组中共检测到36、36、42、40个SSR,SSR类型比较单一,只有单核苷酸和二核苷酸等2种类型,单核苷酸为A和T等2种类型,二核苷酸包括TA和AT等2种类型,其数目比较少。本研究结果与此一致。在本研究中,在‘怀玉山’高山马铃薯叶绿体基因组中共检测到38个SSR位点,其中,单碱基重复有36个,双碱基重复有2个,较少的SSR位点存在表明‘怀玉山’高山马铃薯叶绿体基因组可能不易发生重排。
IR和单拷贝区(SC)边界的膨胀和收缩被认为是被子植物叶绿体全基因组大小变化的主要机制[28],同一属不同品种叶绿体基因组IR/SC边界位置变化也不同[29]。关惜今等[13]研究表明:S. fernandezianum与其野生近缘种(S. phureja、S. palustre、S. etuberosum)叶绿体基因组rps19基因均横跨JLB,S. phureja的ndhF基因横跨JSB,S. fernandezianum、S. palustre、S. etuberosum的ndhF基因均右移,分布在SSC,S. fernandezianum、S. phureja、S. palustre、S. etuberosum的ycf1基因总长度为5 664 bp,均横跨SSC和IRa区域。在本研究中,对‘怀玉山’高山马铃薯及其10个近缘种叶绿体基因组 IR/SC 边界区域的分析结果表明:这些叶绿体基因组的IR都存在扩张或收缩的现象。‘怀玉山’高山马铃薯的rps19基因横跨 JLB,横跨 JLB的左边和右边长度分别为209和69 bp,在JSA,‘怀玉山’高山马铃薯ycf1基因为5 663 bp,左边和右边长度分别为4 541和1 122 bp。
许多植物存在密码子偏好性(CUB),即某一或几种特定密码子频率超过其他同义密码子。密码子偏好性可用来评估基因组中蛋白质编码区(CDS)的密码子使用情况[30]。植物密码子偏好性是物种不断适应外界环境进化所导致的结果,生物获得特定的密码子使用模式以适应起源、进化、自然选择和突变压力等多种因素[31]。影响不同物种中密码子偏好性差异的因素主要有碱基突变、基因表达水平、自然选择等,自然选择和突变压力被认为是2个最重要的因素[32−34]。密码子第3 个碱基的同义突变不能改变氨基酸的类型,但被认为是决定氨基酸类型的重要特征,因此GC3 经常被用作密码子偏向的重要指标[35−36]。本研究发现‘怀玉山’高山马铃薯叶绿体基因组的平均GC 比例为38.38%,GC3为29.60%,更倾向于使用A/T 密码子。RSCU分析结果也证实了这一点。‘怀玉山’高山马铃薯叶绿体基因组中存在A/T 密码子使用偏向,这与大多数高等植物的模式一致[37]。‘怀玉山’高山马铃薯叶绿体基因组平均ENC为47.29,ENC>35的基因有83个,有4个基因的ENC<35,表明‘怀玉山’高山马铃薯叶绿体基因组的密码子偏性较弱。当密码子的使用受到自然选择的影响时,GC3值往往分布在一个较小的范围内,GC12和GC3之间没有显著的相关性[38]。密码子偏好性可以通过调节基因翻译的准确性和效率影响基因表达,基因表达水平越高,密码子偏好性越强[39−40]。通过建立的高低基因表达库,本研究挖掘到‘怀玉山’高山马铃薯叶绿体基因组10个最优密码子,即CGU、AAA、CUU、GUU、GGA、GUA、GGU、UCA、GCU、CCU,说明‘怀玉山’高山马铃薯叶绿体基因组密码子更偏好于以A/U 结尾。筛选到的最优密码子可以用于设计叶绿体基因表达载体,以提高叶绿体基因组中基因的表达水平,也可以利用已知密码子的使用偏好来推测和预测未知基因的表达和功能,可为今后从遗传水平上进行‘怀玉山’高山马铃薯育种改良提供参考。
含有足够信息位点的叶绿体基因组已被证明可有效判断系统发育关系,甚至是在较低的分类学水平下植物之间也有较强的分类学意义,为物种间系统发育的研究提供了新的思路[41]。在本研究中,在茄属中‘怀玉山’高山马铃薯与S. tuberosum ‘Desiree’单独聚为一分支。说明‘怀玉山’高山马铃薯与S. tuberosum ‘Desiree’亲缘关系较近,表明两者同源,推测‘怀玉山’高山马铃薯可能是S. tuberosum ‘Desiree’从美国引种的。
综上所述,本研究测序组装了‘怀玉山’高山马铃薯叶绿体基因组全序列,分析了其编码蛋白基因的密码子使用特点,从高表达优越密码子和高频密码子中选出两者共有的密码子,最终筛选得到了10个叶绿体蛋白编码基因的最优密码子。‘怀玉山’高山马铃薯密码子的偏好性受到突变、选择及其他多方面因素的共同影响,但自然选择的影响更大,这为用基因工程手段改造外源基因密码子,提高其在‘怀玉山’高山马铃薯叶绿体中的表达量提供了参考,也为在分子水平上研究茄科茄属植物的系统进化提供参考。
Chloroplast genome characteristics and codon usage preference of Solanum tuberosum var. cormosus ‘Huaiyushan’
doi: 10.11833/j.issn.2095-0756.20230169
- Received Date: 2023-02-20
- Accepted Date: 2023-11-06
- Rev Recd Date: 2023-10-30
- Available Online: 2023-12-22
- Publish Date: 2024-02-20
-
Key words:
- Solanum tuberosum var. cormosus ‘Huaiyushan’ /
- chloroplast genome /
- sequence characteristics /
- codon preference /
- optimal codons /
- phylogenetic analysis
Abstract:
| Citation: | HONG Senrong, ZHANG Mutong, XU Zilin, et al. Chloroplast genome characteristics and codon usage preference of Solanum tuberosum var. cormosus ‘Huaiyushan’[J]. Journal of Zhejiang A&F University, 2024, 41(1): 92-103. DOI: 10.11833/j.issn.2095-0756.20230169

|

 DownLoad:
DownLoad: