




-
沙棘Hippophae rhamnoides又名醋柳,是胡颓子科Elaeagnaceae沙棘属Hippophae的落叶性灌木[1]。作为药食同源植物的沙棘不仅在食疗、医药、农林牧渔等领域具有较大的经济价值,在水土保持、恢复生物链及防风固沙中也具有极大的生态价值[2-5]。生长过程中沙棘根部会遭受土壤中放线菌、细菌的侵染形成根瘤。部分菌种会在根瘤中高度富集发挥固氮、促生长、抵御逆境胁迫、防止有害病菌侵染等功能[6-8]。传统的微生物研究方法主要以培养基进行分离纯培养,再进而探究其培养特征、显微结构、生理特性等[9]。而自然界中90%以上的微生物为不可培养微生物,且现有培养基与培养技术不适应未知菌群的生长,或部分菌群生长缓慢、丰度较小等情况都会对菌群的多样性评估产生影响[10]。以二代高通量测序为基础的16S rDNA技术通过对编码原核核糖体小亚基rRNA的DNA序列进行测序,不仅克服了传统方法难以获得不可培养菌株的弊端,还能对样品中的物种相对丰度进行排序,并分析各群组样品中发挥重要作用的优势物种,解析样品中微生物之间的相互作用。该技术对研究沙棘根瘤内生菌微生物多样性与环境关系以及微生物资源的开发利用有重要的理论和现实意义[11-16]。
本研究通过16S rRNA测序技术对沙棘根瘤内生菌进行物种注释、分类学分析、α多样性分析、β多样性分析、组间差异显著性分析,比较高通量测序和纯培养方法的差异与优劣,为发掘具有应用价值的根瘤内生菌资源提供科学依据。
-
采样地为内蒙古自治区巴彦淖尔市磴口县中国林业科学研究院沙漠林业实验中心试验林场(40°29′34″N,106°74′06″E)。该研究区海拔为1 054 m,年平均气温为7.4 ℃。2020年7月,选取人为干扰因素较少的沙漠边缘地带采集沙棘根瘤样品。在每个样地10 m×10 m的区域内用网格法定9个点,运用梅花形采样法在边角及中心共5个点分别采集根瘤样品并进行混合,共设计6组重复样,分别命名为M1、M2、M3、M4、M5、M6。
-
依据文献[17-18]的方法进行修改,使其更加适宜沙棘根瘤内生菌的分离。详细步骤如下:选取新鲜饱满的根瘤,冲洗掉土粒泥沙,将根瘤团用解剖刀分割成带有单柄的瘤瓣,用纱布包裹,先用体积分数为95%的酒精溶液浸泡30 s,再用体积分数为10%的次氯酸钠溶液表面灭菌5 min,取出后用无菌水冲洗数次。在灭菌滤纸上,用无菌解剖刀先切取根瘤头部,再将其均分成2~3份薄片,置于固体培养基中28 ℃恒温暗处静置培养。根据相关研究,本研究选取BAP[19]、S[20]、JA[19]、高氏一号培养基[19]进行分离培养。
-
提取纯培养的沙棘根瘤内生菌DNA后,对16S rDNA全长进行PCR扩增。序列引物采用YU等[21]设计的细菌通用引物(引物序列27F:5′-AGAGTTTGATCMTGGCTCAG-3′,1492R:5′-GGYTACCTTGTTACGACTT-3′),PCR总反应体系为50 μL,包括10×缓冲液(KOD buffer) 5 μL、2 mmol·L−1三磷酸脱氧核糖核苷酸混合液(dNTPs) 5 μL、基因组DNA (genomic DNA) 1 μL、上游引物(forward primer) (10 μm) 1 μL、下游引物(reverse primer) (10 μm) 1 μL、DNA聚合酶(KOD DNA polymerase) 1 μL、超纯水(ddH2O) 36 μL。PCR反应程序:94 ℃预变性 3 min,94 ℃变性 30 s,58 ℃退火 30 s,72 ℃延伸1 min,35个循环,最后72 ℃延伸10 min。用质量分数为1%的琼脂糖凝胶电泳,确定有特异扩增后,进行PCR产物回收和测序注释,并参考文献[22-25]进行比对校验。
-
提取沙棘根瘤总DNA后,根据16S rDNA保守区设计引物(引物序列335F:5′-CADACTCCTACGGGAGGC-3′,769R:5′-ATCCTGTTTGMTMCCCVCRC-3′),在引物末端加上测序接头,便于建库时添加能区分样本的碱基序列的条码/索引(barcode/index)。再进行PCR扩增并对其产物进行紫外分光光度计定量及混样、过柱纯化和均一化形成测序文库,建好的文库先进行文库质检,质检合格的文库用Illumina HiSeq 2500进行测序[26]。高通量测序得到的原始图像数据文件,经碱基识别分析转化为原始测序序列,结果以FASTQ (简称为fq)文件格式存储[27]。
-
首先使用 Trimmomatic v.0.33软件[28],对测序得到的原始测序序列进行过滤;其次使用cutadapt 1.9.1软件进行引物序列的识别与去除,得到不包含引物序列的高质量测序序列;然后使用FLASH v1.2.7软件[29],按照最小重叠(overlap)长度为10 bp、重叠区允许的最大错配比率为0.2的要求,对每个样品高质量的一小段短的基因测序片段(reads)进行拼接,得到的拼接序列即原始序列质控后的高质量测序序列(clean reads);最后使用UCHIME v4.2软件[30],鉴定并去除嵌合体序列,得到最终有效数据。使用Usearch软件对reads在97.0%的相似度水平下进行聚类,获得分类操作单元(OTU)[31],以测序所有序列数的0.005%作为阈值过滤OTU[32]。以SILVA (
http://www.arb-silva.de/ )为参考数据库使用朴素贝叶斯分类器对特征序列进行分类学注释,可得到每个特征对应的物种分类信息,进而在各水平(门、纲、目、科、属、种)统计样品群落组成,利用QIIME软件生成不同分类水平上的物种丰度表,再利用R语言工具绘制样品分类学水平下的群落结构图[33]。使用QIIME软件对样品α多样性进行评估和t检验(显著性水平为0.01)。利用Mothur v1.30软件和R语言工具包绘制稀释曲线。基于独立OTU,采用加权分析方法和Bray-Curtis算法,使用QIIME软件进行非加权组平均法(UPGMA)分析,比较各组样品间的物种差异。 -
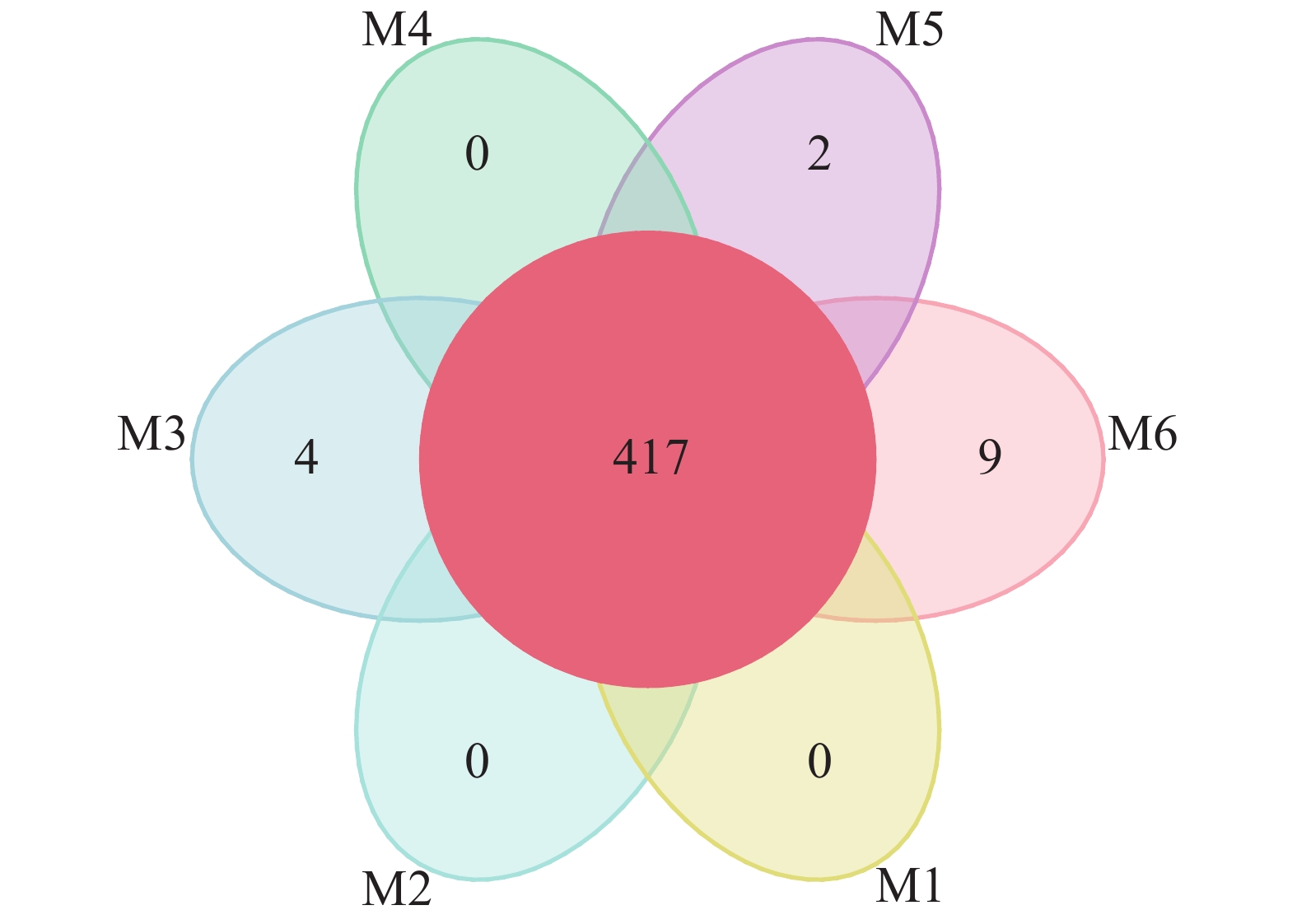
使用Usearch软件对clean reads在97.0%的相似度水平下进行聚类,共计获得651个OTU。各样品OTU个数分布较为均匀,样品M1~M6分别为551、583、579、518、593、589个。如图1所示:6组样品中共有的OTU数为417个。M3、M5、M6中分别有4、2、9个特有的OTU,为样品特有OTU,非单个样品特有或所有样品间共有的OTU在图1未做展示。从整体来看,不同地点的各样品间的OTU差异性远小于共性,说明采样方法设计合理。
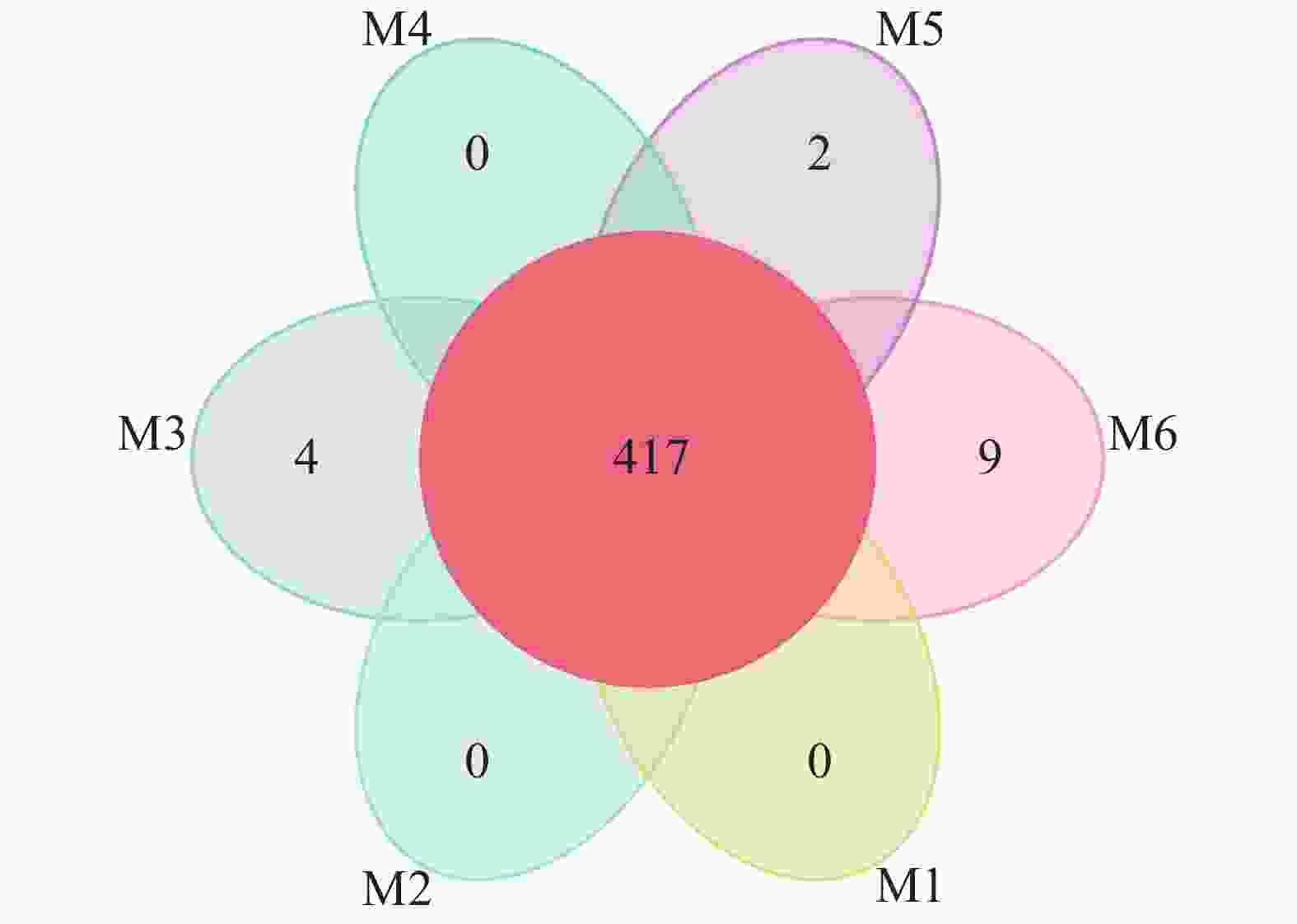
图 1 沙棘(M1~M6)根瘤样品分类操作单元(OTU)花瓣图
Figure 1. Petal image of operational taxonomic unit (OTU) of H. rhamnoides root nodule sample (M1-M6)
-
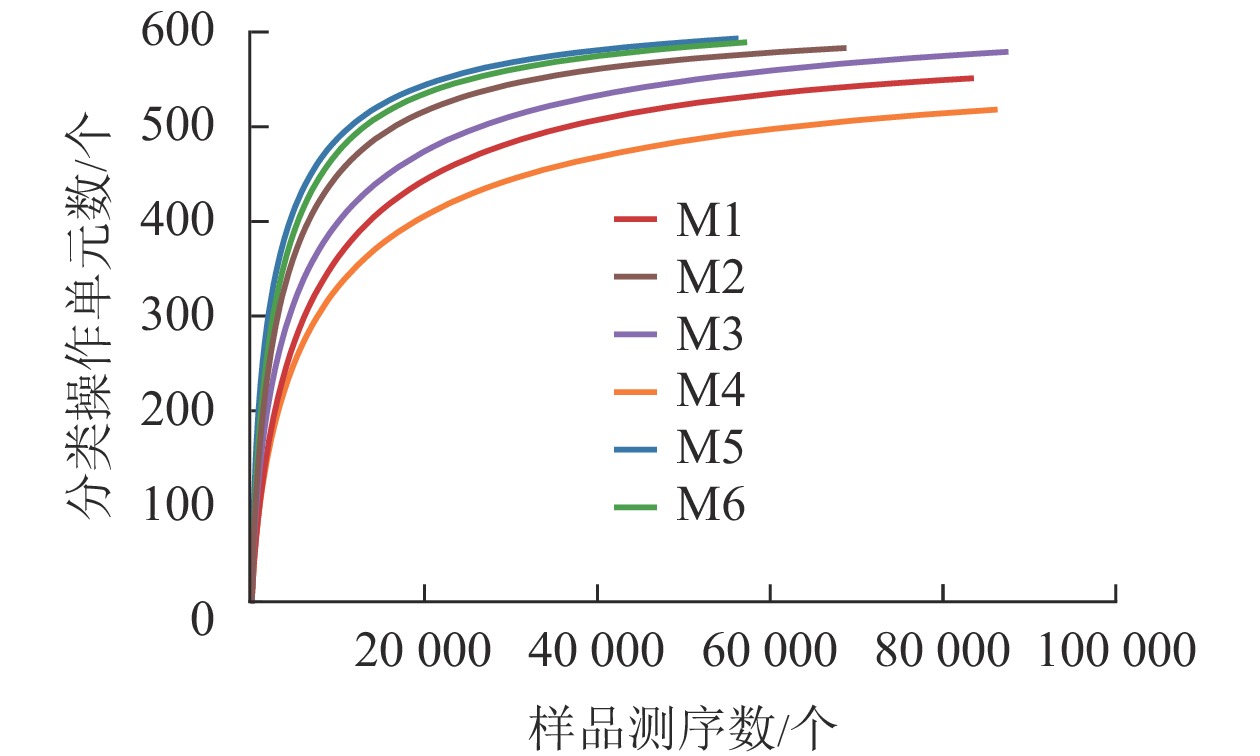
对6组样品测序共获得 810 039对reads,双端reads质控、拼接后共产生617 188条clean reads。其中质量≥20的碱基占总碱基数的比例(Q20)为98.7%,质量≥30的碱基占总碱基数的比例(Q30)为95.4%,表明测序质量较好。从图2可见:各样品稀释性曲线趋向平缓,表明在持续抽样下新物种出现的速率逐渐趋于平缓,此环境中物种数量不会随测序数量的增加而显著增多[34],说明取样合理,能较好体现6组样品中根瘤内生菌的多样性,可以进行数据分析。M5的Shannon和Simpson指数最大(表1),说明物种多样性最高。同理,M4的物种多样性最低。物种丰度方面M5与M6差别不大,均有较高水平。M4根瘤样品的物种丰度最低。样点的Shannon指数平均为4.24,Simpson指数平均为0.70,Ace指数平均为585.79,Chao1指数平均为595.47,样本文库平均覆盖率为99.95%。说明采样地的沙棘根瘤内生菌的物种丰富且多样性较大,各物种分配相对均匀,其微生物物种信息得到了充分体现。
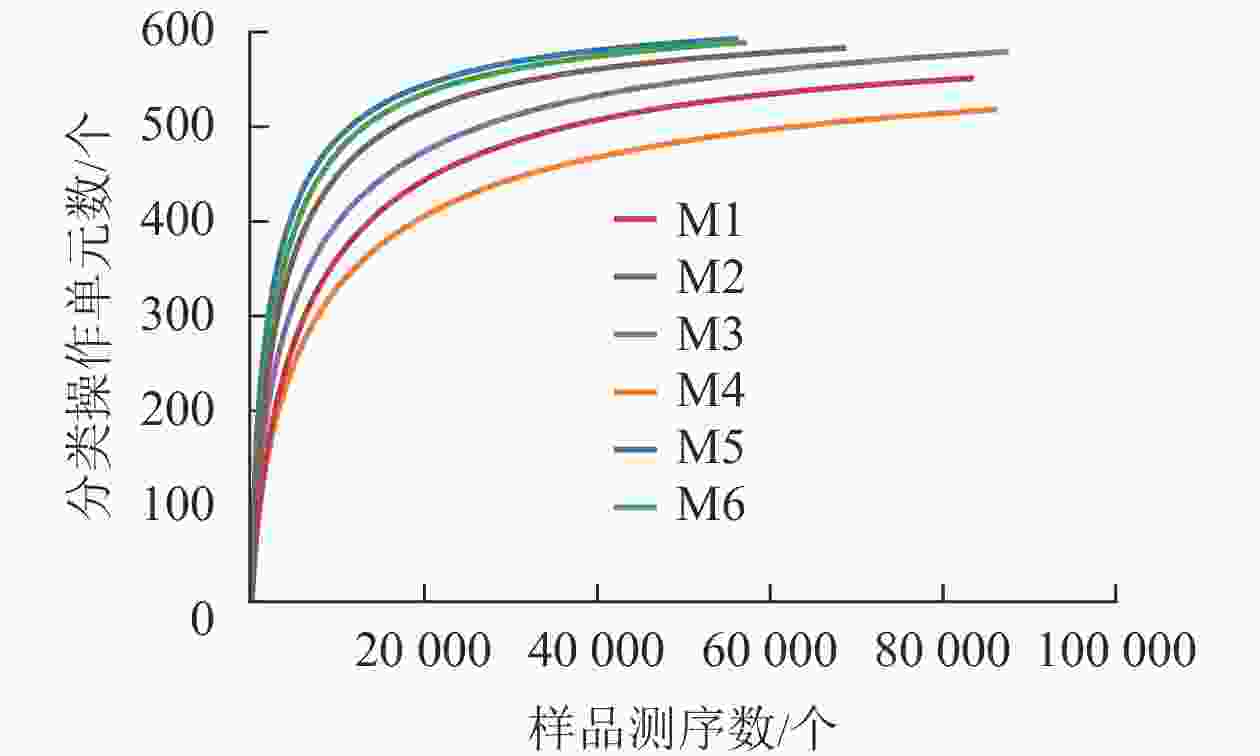
图 2 各样品稀释性曲线
Figure 2. Dilution curve of each sample
表 1 各组样品的α多样性指数
Table 1. Alpha diversity index for each group of samples
样品 Shannon
指数Simpson
指数Ace
指数Chao1
指数覆盖
率/%M1 2.53 0.47 568.45 598.57 99.95 M2 4.73 0.79 595.09 600.60 99.95 M3 4.28 0.75 600.58 607.45 99.95 M4 2.52 0.44 542.32 543.52 99.94 M5 6.58 0.95 605.66 610.71 99.94 M6 4.82 0.77 602.63 611.97 99.94 平均 4.24 0.70 585.79 595.47 99.95 -
通过传统分离方法从BAP、JA、S、高氏一号培养基中得到纯培养菌株96株。所有菌株均可传代培养,但菌株之间培养周期差异较大,培养周期在1~30 d呈离散型分布。对各菌株进行分子鉴定,共有4门8纲8目13科19属。在门的分类水平分别为变形菌门Proteobacteria、放线菌门Actinobacteria、厚壁菌门Firmicutes和柔膜菌门Tenericutes。在属的分类水平上,96株菌分属于支原体属Mycoptasma 1株、慢生根瘤菌属Bradyrhizobim 6株、土壤杆菌属Agrobacterium 7株、肠杆菌属Enterobacter 6株、小坂菌属Kosakonia 8株、柠檬酸杆菌属Citrobacter 1株、约克氏菌属Yokenella 1株、欧文氏菌属Erwinia 1株、克罗诺杆菌属Cronobacter 2株、泛菌属Pantoea 1株、莫拉菌属Moraxella 1株、贪噬菌属Variovorax 1株、草螺菌属Herbaspirillum 1株、假单胞菌属Pseudomonas 5株、链霉菌属Streptomyces 14株、小单孢菌属Micromonospora 1株、短杆菌属Brevibacterium 6株、葡萄球菌属Straphylococcus 1株和芽孢杆菌属Bacillus 32株。其中,优势门为变形菌门和厚壁菌门,优势属为芽孢杆菌属和链霉菌属。
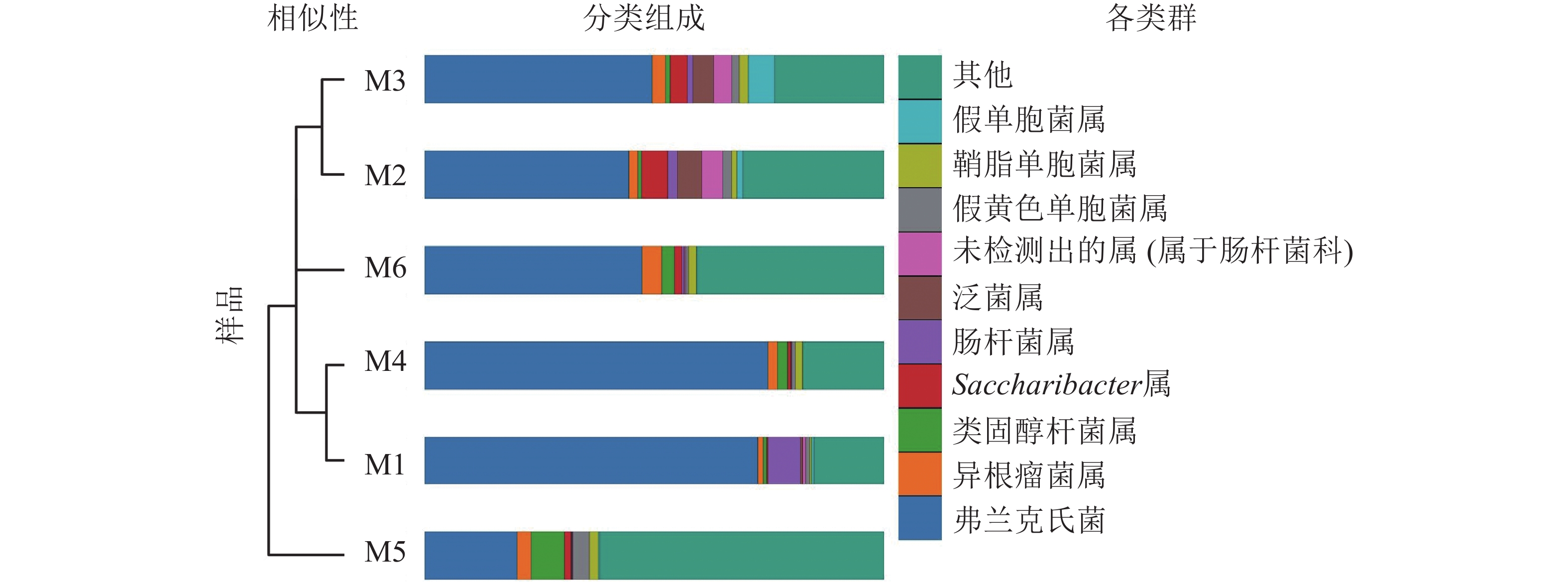
高通量测序分析发现:6组样品共有14门34纲89目148科314属。将相对丰度大于0.1%的门与相对丰度前10的属进行汇总(图3、表2、表3)发现:在门的分类水平上,6组样品中相对丰度较高的主要为放线菌门和变形菌门,两者相对丰度之和为87.5%~97.1%。其次为拟杆菌门Bacteroidetes、杆菌门Patescibacteria、厚壁菌门、酸杆菌门Acidobacteria。在属的分类水平上,弗兰克氏菌属Frankia占绝对优势,相对丰度为20.12%~74.81%,平均相对丰度为51.49%。其次为根瘤菌属Rhizobium、类固醇杆菌属Steroidobacter、糖单孢菌属Saccharimonadales、肠杆菌属、泛菌属、欧文氏菌属、假黄色单胞菌属Pseudoxanthomonas、鞘脂单胞菌属Sphingomonas、假单胞菌属、固氮弓菌属Azoarcus、伯克氏菌属Burkholderia、芽单胞菌属Blastomonas、聚集杆菌属Congregibacter、拉恩氏菌属Rahnella、鞘氨醇菌属Chitinophaga、独岛杆菌属Dokdonella、普雷沃氏菌属Prevotella、链霉菌属、Microtrichales属。
表 2 沙棘微生物区系门水平的相对分布
Table 2. Relative abundance of microbiota taxa at the level of phylum
分类 6组样品在门水平的相对丰度/% M1 M2 M3 M4 M5 M6 放线菌门 73.55 47.56 51.24 76.09 27.73 57.68 变形菌门 22.38 41.91 41.63 21.01 60.35 29.82 拟杆菌门 0.89 1.42 1.30 0.40 2.18 4.42 杆菌门 0.31 5.66 3.89 0.73 1.42 1.72 厚壁菌门 2.18 2.74 1.30 1.21 2.18 4.42 酸杆菌门 0.15 0.26 0.42 0.18 1.16 0.53 其他 0.54 0.45 0.22 0.38 0.75 0.60 表 3 沙棘微生物区系属水平的相对分布
Table 3. Relative abundance of microbiota taxa at the level of genus
分类 6组样品在属水平的相对丰度/% M1 M2 M3 M4 M5 M6 弗兰克氏菌属 72.62 44.45 49.50 74.81 20.12 47.41 根瘤菌属 1.17 1.97 2.89 2.04 3.13 4.13 类固醇杆菌属 0.73 0.83 1.05 2.20 7.19 2.87 糖单孢菌属 0.28 5.61 3.85 0.71 1.41 1.68 肠杆菌属 6.93 2.19 1.05 0.08 0.22 0.40 泛菌属 0.63 5.19 4.69 0.01 0.05 0.11 欧文氏菌属 0.60 4.66 3.77 0.10 0.18 0.23 假黄色单胞菌属 0.85 1.98 1.67 0.75 3.56 0.68 鞘脂单胞菌属 0.39 1.06 2.10 1.55 2.10 1.64 假单胞菌属 0.51 1.27 5.60 0.03 0.21 0.09 其他 15.29 30.79 23.83 17.72 61.83 40.76 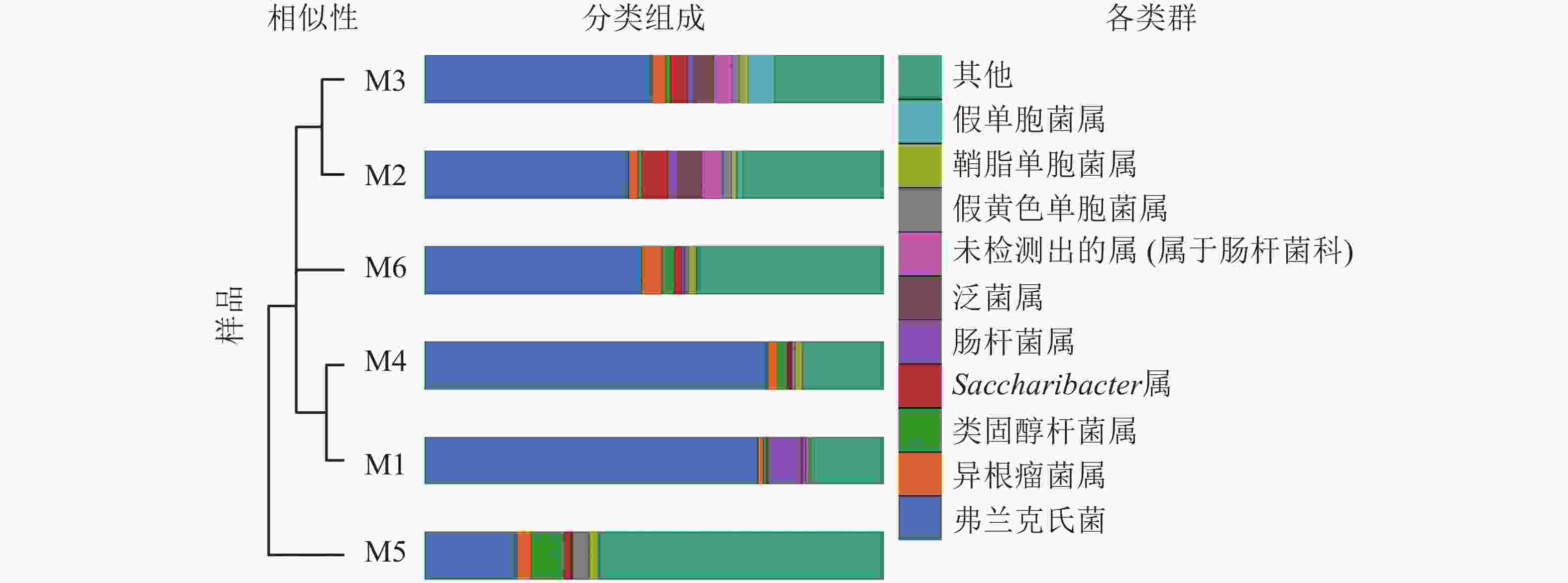
图 3 6组根瘤样品的非加权组平均法(UPGMA)聚类树与物种分布柱状图
Figure 3. UPGMA clustering tree and the species distribution histogram of the six groups of nodule samples are combined drawing
在门、纲、目、科、属的各分类单元中,高通量测序的检测灵敏度(高通量测序/纯培养)依次是纯培养方法的3.50、4.25、11.20、11.38和16.53倍。在门水平上,纯培养菌株中占比较高的厚壁菌门在高通量测序中占比并不高。在属水平上,纯培养菌株中占比较高的芽孢杆菌属和链霉菌属皆在高通量测序中占比很低。该对比结果差异性较大,说明高通量测序在微生物多样性分析中占据优势地位,要优于纯培养方法。同时也说明,沙棘根瘤内共生细菌群落结构更为复杂,群落更为稳定。
-
在运用传统方法分离纯培养微生物时,共分离纯培养菌株96株,分属于4门8纲8目13科19属,未获得弗兰克氏菌属的菌株,可能是培养基中弗兰克氏菌属的菌株生长缓慢,易被其他菌群取代,因此仍需探索新的培养基与培养方法以遏制根瘤中其他菌株的繁殖。在微生物多样性分析中,由于环境中的微生物复杂多样,各环境之间组成差异较大,通常采用非加权方法进行分析。该方法简单易操作,主要考虑物种的有无,但未考虑物种的丰度,所以采用非加权的方法难以区别各样品间的差异。
高通量测序分析共检测到14门34纲89目148科314属。在门、纲、目、科、属的各分类单元中,高通量测序的检测灵敏度(高通量测序/纯培养)依次是纯培养方法的3.50、4.25、11.20、11.38和16.53倍。与纯培养获得的菌株相比,高通量测序分析结果更加完整地揭示了沙棘根瘤内生菌的微生物多样性。高通量测序表明:在门的分类水平上,样品中相对丰度较高的主要为放线菌门和变形菌门,两者相对丰度之和为87.5%~97.1%。在属的分类水平上,弗兰克氏菌属占绝对优势,相对丰度为20.12%~74.81%,平均相对丰度为51.49%。
-
张爱梅等[35]和刘志强等[36]分别对甘肃榆中、辽宁通辽、内蒙古赤峰等地沙棘根瘤内生菌微生物多样性做过类似研究,其高通量测序所得的微生物多样性高于本研究结果,说明沙棘根瘤内生菌微生物多样性受地理位置、土壤成分、气候条件、宿主种类及生长环境等多种因素的影响。本研究的沙棘取样于内蒙古乌兰察布沙漠边缘地带,采样地荒漠化土壤与干旱少雨气候对内生菌多样性有特别影响。
属于非豆科Leguminosae植物的沙棘根瘤共生固氮体系是以弗兰克氏菌属为主导的[37]微生物—微生物—植物互作体系。高通量测序分析显示:弗兰克氏菌属所占比例较高,然而本次传统方法分离却未得到纯培养菌株,这可能是由于培养基中缺乏某种信号物质或与其他菌属竞争存在劣势导致的,建议添加制霉菌素、萘啶酮酸和放线菌酮抑制其他菌群的繁殖[18]。非豆科植物结瘤固氮过程,单一属的菌株难以完成此任务。有研究[38]表明:纯培养分离的贪噬菌属是复杂微生物组中维持根生长的核心菌属,并且具有产生和降解生长素的能力,是细菌—细菌—植物通讯网络的关键角色。小单孢菌是植物益生菌,在促进植物生长的同时还可以分泌细胞壁降解酶促进细胞壁的降解,进而便于弗兰克氏菌的侵染[39-40],但是小单孢菌的快速繁殖也对弗兰克氏菌的生长起到抑制作用。沙棘作为胡颓子科植物,根部结瘤侵染方式为细胞间侵入。研究[41]表明:草螺旋菌属Spirillum、慢生根瘤菌属、肠杆菌属的相关细菌与弗兰克氏菌存在负相关性(即抑制关系),以上3个菌属均在豆科、禾本科Poaceae植物中发挥固氮相关的重要作用,但在胡颓子科中此类细菌与弗兰克氏菌属相互作用的机制尚未明确。
Construction of endophytic strain bank of seabuckthorn nodule and an analysis of microbial diversity
-
摘要:
目的 沙棘Hippophae rhamnoides根瘤中拥有丰富的微生物资源,探究根瘤内生菌微生物多样性,比较高通量测序与纯培养方法的优劣。 方法 运用16S rRNA高通量测序技术探究沙棘根瘤内生菌相对丰度和多样性差异,并对根瘤内生菌进行分离纯培养,以完成内生菌株库初步构建。 结果 ①传统分离方法得到纯培养菌株96株,共4门8纲8目13科19属。在门分类水平上,相对丰度较高的主要为变形菌门Proteobacteria和厚壁菌门Firmicutes。②高通量测序分析6组样品(M1~M6)中共有14门34纲89目148科314属。在门分类水平上,相对丰度较高的主要为放线菌门Actinobacteria和变形菌门,两者相对丰度之和为87.50%~97.10%。在属的分类水平上,弗兰克氏菌属Frankia占绝对优势,相对丰度为20.12%~74.81%,平均相对丰度为51.49%。③高通量测序与纯培养方法的结果有明显差异,在科和属的分类单元上差异较大。 结论 2种方法都体现沙棘根瘤内生菌的多样性,但纯培养方法仅能够分离到部分内生菌,难以评估物种组成及相对丰度。高通量测序分析能够更为全面地反映多样性信息,且为优化纯培养条件而分离特定物种奠定基础。图3表3参41 -
关键词:
- 根瘤内生菌 /
- 微生物多样性 /
- 16S rDNA高通量测序 /
- 菌株库构建 /
- 沙棘
Abstract:Objective This study is aimed to conduct an investigation of the microbial diversity endophytic bacteria in rhizobia of Hippophae rhamnoides which play an important role in nitrogen fixation and plant growth and research the construction of rhizobia endophytes. Method With the employment of 16S rRNA high-throughput sequencing technology, an exploration was conducted of the relative abundance and diversity of endophytes in Ulange wood rhizobia of Mongolian H. rhamnoides in Dengkou County before the endophytes were isolated and purely cultured to complete the construction of endophytic strains library. Result (1) A total of 96 pure culture strains were obtained by traditional isolation, with 4 phyla, 8 classes, 8 orders, 13 families, and 19 genera and Proteobacteria and Firmicutes are of relatively higher relative abundances in terms of phyla classification. (2) With an analysis of 6 groups of samples (M1−M6) consisting of 14 phyla, 34 classes, 89 orders, 148 families, and 314 genera by high-throughput sequencing, Actinomycota and Proteobacteria have higher relative abundance, and the sum of the relative abundance of the two is between 87.50%−97.10% in terms of phylum classification. As for the taxonomic level of the genus, the genus Frankia occupies an absolute advantage with the relative abundance being between 20.12% and 74.81%, and the average relative abundance being 51.49%. With M5 having the largest Shannon and Simpson indexes and M4 having the lowest species diversity, both M5 and M6 have higher levels of species abundance whereas M4 species has the lowest abundance. (3) The results of high-throughput sequencing and pure culture methods are significantly different, especially in the taxa of families and genera. Conclusion Both methods reflected the diversity of endophytes in sea buckthorn rhizobia, but the pure culture method could only isolate some endophytes, and it was difficult to evaluate the species composition and relative abundance. Also, high-throughput sequencing analysis could reflect the diversity better and lay the foundation for optimizing pure culture conditions for the isolation of specific species. [Ch, 3 fig. 3 tab. 41 ref.] -
图 1 沙棘(M1~M6)根瘤样品分类操作单元(OTU)花瓣图
Figure 1 Petal image of operational taxonomic unit (OTU) of H. rhamnoides root nodule sample (M1-M6)
图 3 6组根瘤样品的非加权组平均法(UPGMA)聚类树与物种分布柱状图
Figure 3 UPGMA clustering tree and the species distribution histogram of the six groups of nodule samples are combined drawing
表 1 各组样品的α多样性指数
Table 1. Alpha diversity index for each group of samples
样品 Shannon
指数Simpson
指数Ace
指数Chao1
指数覆盖
率/%M1 2.53 0.47 568.45 598.57 99.95 M2 4.73 0.79 595.09 600.60 99.95 M3 4.28 0.75 600.58 607.45 99.95 M4 2.52 0.44 542.32 543.52 99.94 M5 6.58 0.95 605.66 610.71 99.94 M6 4.82 0.77 602.63 611.97 99.94 平均 4.24 0.70 585.79 595.47 99.95  下载: 导出CSV
下载: 导出CSV
表 2 沙棘微生物区系门水平的相对分布
Table 2. Relative abundance of microbiota taxa at the level of phylum
分类 6组样品在门水平的相对丰度/% M1 M2 M3 M4 M5 M6 放线菌门 73.55 47.56 51.24 76.09 27.73 57.68 变形菌门 22.38 41.91 41.63 21.01 60.35 29.82 拟杆菌门 0.89 1.42 1.30 0.40 2.18 4.42 杆菌门 0.31 5.66 3.89 0.73 1.42 1.72 厚壁菌门 2.18 2.74 1.30 1.21 2.18 4.42 酸杆菌门 0.15 0.26 0.42 0.18 1.16 0.53 其他 0.54 0.45 0.22 0.38 0.75 0.60
下载: 导出CSV
表 3 沙棘微生物区系属水平的相对分布
Table 3. Relative abundance of microbiota taxa at the level of genus
分类 6组样品在属水平的相对丰度/% M1 M2 M3 M4 M5 M6 弗兰克氏菌属 72.62 44.45 49.50 74.81 20.12 47.41 根瘤菌属 1.17 1.97 2.89 2.04 3.13 4.13 类固醇杆菌属 0.73 0.83 1.05 2.20 7.19 2.87 糖单孢菌属 0.28 5.61 3.85 0.71 1.41 1.68 肠杆菌属 6.93 2.19 1.05 0.08 0.22 0.40 泛菌属 0.63 5.19 4.69 0.01 0.05 0.11 欧文氏菌属 0.60 4.66 3.77 0.10 0.18 0.23 假黄色单胞菌属 0.85 1.98 1.67 0.75 3.56 0.68 鞘脂单胞菌属 0.39 1.06 2.10 1.55 2.10 1.64 假单胞菌属 0.51 1.27 5.60 0.03 0.21 0.09 其他 15.29 30.79 23.83 17.72 61.83 40.76
下载: 导出CSV
-
[1] 兰士波. 中国沙棘良种选育及遗传改良研究进展与展望[J]. 经济林研究, 2011, 29(3): 102 − 106. LAN Shibo. Research progress and prospects of selection and breeding of superior Chinese species in Hippophae and its genetic improvement [J]. Non-wood For Res, 2011, 29(3): 102 − 106. [2] LIU Yong, QIU Yuping, ZHANG Ling, et al. Dormancy breaking and storage behavior of Garcinia cowa Roxb. (Guttiferae) seeds: implications for ecological function and germplasm conservation [J]. J Integrative Plant Biol Formerly Acta Bot Sin, 2005, 47(1): 38 − 49. [3] VIBHA B, NEELAM G. Importance of exploration of microbial biodiversity [J]. ISCA J Biol Sci, 2012, 1(3): 78 − 83. [4] BERNER D, VIERNSTEIN H. Effect of protective agents on the viability of Lactococcus lactis subjected to freeze-thawing and freeze-drying [J]. Sci Pharm, 2006, 74(3): 137 − 149. [5] BOUMAHDI M, MARY P, HORNEZ J P. Influence of growth phases and desiccation on the degrees of unsaturation of fatty acids and the survival rates of rhizobia [J]. J Appl Microbiol, 2010, 87(4): 611 − 619. [6] CHIAN R C, QUINN P. Fertility Cryopreservation: Cryopreservation of Sperm and Testicular Tissue[M]. Cambridge: Cambridge University Press, 2010: 160 − 162. [7] ZHOU Yiqin, DROUIN P, LAFRENIÈRE C. Effects on microbial diversity of fermentation temperature (10 ℃ and 20 ℃), long-term storage (5 ℃), and subsequent warming on corn silage [J]. Asian-Australas J Anim Sci, 2019, 32(10): 1528 − 1539. [8] SMIRNOVA D V, ZALOMOVA L V, ZAGAINOVA A V, et al. Cryopreservation of the human gut microbiota: current state and perspectives [J]. Int J Med Microbiol, 2019, 309(5): 259 − 269. [9] 刘泉成. 玉米根际微生物群落特征分析及生防菌筛选[D]. 北京: 中国农业科学院, 2018. LIU Quancheng. Analyzing Composition of Bacterial Community and Screening of Biocontrol Bacteria Strains in Maize Rhizosphere [D]. Beijing: Chinese Academy of Agricultural Sciences, 2018. [10] 许来鹏, 万鲜花, 孙向丽, 等. 畜禽粪肥和秸秆还田对玉米根际微生物群落结构的影响[J]. 生物技术通报, 2020, 36(9): 142 − 151. XU Laipeng, WAN Xianhua, SUN Xiangli, et al. Effects of livestock manure and straw returning to field on microbial community structure around maize rhizosphere [J]. Biotechnol Bull, 2020, 36(9): 142 − 151. [11] 谢宪, 梁军, 张铭, 等. 赤松枯梢病叶内生真菌多样性研究[J]. 生物技术通报, 2020, 36(2): 119 − 125. XIE Xian, LIANG Jun, ZHANG Ming, et al. Endophytic fungi diversity in the needles of Pinus densiflora with Sphaeropsis sapinea [J]. Biotechnol Bull, 2020, 36(2): 119 − 125. [12] 马慧媛. 微生物菌剂施用对设施茄子根际土壤养分和细菌群落多样性的影响[J]. 微生物学通报, 2020, 47(1): 140 − 150. MA Huiyuan. Effects of microbial agents on nutrient and bacterial community diversity in rhizosphere soil of eggplant cultivated in facilities [J]. Microbiol China, 2020, 47(1): 140 − 150. [13] 牛世全, 龙洋, 李海云, 等. 应用Illumina MiSeq 高通量测序技术分析河西走廊地区盐碱土壤微生物多样性[J]. 微生物学通报, 2017, 44(9): 63 − 74. NIU Shiquan, LONG Yang, LI Haiyun, et al. Microbial diversity in saline alkali soil from Hexi Corridor analyzed by Illumina MiSeq high-throughput sequencing system [J]. Microbiol China, 2017, 44(9): 63 − 74. [14] 邓振山, 商荣芳, 陈凯凯, 等. 陕北地区提高石油采收率菌株筛选及其降解性能评价[J]. 生物技术通报, 2019, 35(2): 73 − 79. DENG Zhenshan, SHANG Rongfang, CHEN Kaikai, et al. Screening of strains for enhancing oil recovery in Northern Shaanxi and evaluation of their degradabilities [J]. Biotechnol Bull, 2019, 35(2): 73 − 79. [15] 杨溢, 黄美, 张琛, 等. 16s rRNA 高通量测序技术在人类医学中的应用进展[J]. 亚洲临床医学杂志, 2019, 1(1): 16 − 17. YANG Yi, HUANG Mei, ZHANG Chen, et al. Application progress of 16s rRNA high-throughput sequencing technology in human medicine [J]. Asian J Clin Med, 2019, 1(1): 16 − 17. [16] 王志伟, 纪燕玲, 陈永敢. 植物内生菌研究及其科学意义[J]. 微生物学通报, 2015, 42(2): 349 − 363. WANG Zhiwei, JI Yanling, CHEN Yonggan. Studies and biological significances of plant endophytes [J]. Microbiol China, 2015, 42(2): 349 − 363. [17] 徐瑞瑞. 沙棘弗兰克氏和非弗兰克氏放线菌的分离、培养及分子鉴定[D]. 北京: 中国林业科学研究院, 2011. XU Ruirui. The Isolation, Cuture and Molecular Identification of Fraknkia and Non-Frankia Sctionayetec form Hipooha rehanmde Root Nodules [D]. Beijing: Chinese Academy of Forestry, 2011. [18] 李志真. 福建弗兰克氏菌Frankia研究[D] . 福州: 福建农林大学, 2002. LI Zhizhen. The Studies on Actinomycetes-Frankia in Fujian [D]. Fuzhou: Fujian Agriculture and Forestry University, 2002. [19] 刁治民, 陈克龙, 王文颖. 固氮微生物学[M]. 北京: 科学出版社, 2014: 169 − 188. DIAO Zhimin, CHEN Kelong, WANG Wenying. Nitrogen Fixation Microbiology[M]. Beijing: Science Press, 2014: 169 − 188. [20] 李利坤. 沙棘根瘤菌的分离鉴定及根瘤菌对植株生长发育的影响[D]. 长春: 吉林农业大学, 2018. LI Likun. Isolation and Identification of Rhizobium from Seabuckthorn and Effects of Rhizobium on the Growth and Development of Plants [D]. Changchun: Jilin Agricultural University, 2018. [21] YU Jie, ZHOU Xiaofeng, YANG Suijuan, et al. Design and application of specific 16S rDNA-targeted primers for assessing endophytic diversity in Dendrobium officinale using nested PCR-DGGE [J]. Appl Microbiol Biotechnol, 2013, 97(22): 9825 − 9836. [22] 周小倩. 黑龙江省不同经度耕作生态系统土壤微生物群落结构研究[D]. 哈尔滨: 哈尔滨师范大学, 2016. ZHOU Xiaoqian. Study on the Microbial Community Structure in Farming Ecosystem in Different Longitudes of Heilongjiang Province [D]. Harbin: Harbin Normal University, 2016. [23] 阮继生, 黄英. 放线菌快速鉴定与系统分类[M]. 北京: 科学出版社, 2011: 60 − 62. RUAN Jisheng, HUANG Ying. Rapid Identification and Systematic Classification of Actinomycetes[M]. Beijing: Science Press, 2011: 60 − 62. [24] 董秀珠, 蔡妙英. 常见细菌系统鉴定手册[M]. 北京: 科学出版社, 2001: 353 − 390. DONG Xiuzhu, CAI Miaoying. The Identification Manual of Common Bacteria System[M]. Beijing: Science Press, 2001: 353 − 390. [25] 李振高, 骆永明, 滕应. 土壤与环境微生物研究法[M]. 北京: 科学出版社, 2008: 146 − 201. LI Zhengao, LUO Yongming, TENG Ying. Soil and Environmental Microbiology Research Method[M]. Beijing: Science Press, 2008: 146 − 201. [26] 刘欣, 李志英, 刘瑞瑞, 等. 大豆不同生育期根际土壤细菌群落结构的变化[J]. 广西植物, 2018, 38(10): 107 − 114. LIU Xin, LI Zhiying, LIU Ruirui, et al. Changes of bacterial flora structure in rhizosphere soil of soybean at different growth stages [J]. Guihaia, 2018, 38(10): 107 − 114. [27] 阚望, 李成云, 许姗姗, 等. 西双版纳热带雨林植物叶际细菌群落研究[J]. 西南农业学报, 2020, 33(3): 631 − 636. KAN Wang, LI Chengyun, XU Shanshan, et al. Plant phyllosphere bacteria community in Xishuangbanna tropic rainforest in China [J]. Southwest China J Agric Sci, 2020, 33(3): 631 − 636. [28] BOLGER A M, LOHSE M, USADEL B, et al. Trimmomatic: a flexible trimmer for Illumina sequence data [J]. Bioinformatics, 2014, 30(15): 2114 − 2120. [29] MAGO T, SALZBERG S L. FLASH: fast length adjustment of short reads to improve genome assemblies [J]. Bioinformatics, 2011, 27(21): 2957 − 2963. [30] EDGAR R C, HAAS B J, CLEMENTE J C, et al. UCHIME improves sensitivity and speed of chimera detection [J]. Bioinformatics, 2011, 27(16): 2194 − 2200. [31] EDGAR R C. UPARSE: highly accurate OTU sequences from microbial amplicon reads [J]. Nat Methods, 2013, 10(10): 996 − 998. [32] BOKULICH N A, SUBRAMANIAN S, FAITH J J, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing [J]. Nat Methods, 2013, 10(1): 57 − 59. [33] 李怡, 余林, 况小宝. 未覆盖雷竹林土壤细菌群落剖面分布特征[J]. 南方林业科学, 2019, 47(4): 1 − 5. LI Yi, YU Lin, KUANG Xiaobao. The profile distribution characteristics of soil bacterial community in uncovered Phyllostachys praecox forest [J]. Southern China For Sci, 2019, 47(4): 1 − 5. [34] WANG Yu, SHENG Huafang, HE Yan, et al. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags [J]. Appl Environ Microbiol, 2012, 78(23): 64 − 82. [35] 张爱梅, 韩雪英, 王嘉, 等. 马衔山中国沙棘根瘤内共生细菌多样性研究[J]. 生态学报, 2019, 39(1): 298 − 305. ZHANG Aimei, HAN Xueying, WANG Jia, et al. Diversity of endophytic bacteria in root nodules of Hippophae rhamnoides in the Maxian Mountains [J]. Acta Ecol Sin, 2019, 39(1): 298 − 305. [36] 刘志强, 张利平. 沙棘和沙枣根瘤中固氮放线菌物种多样性研究[J]. 安徽农业科学, 2010, 38(11): 33 − 36. LIU Zhiqiang, ZHANG Liping. Study on species diversity of nitrogen-fixing actinomycetes in Hippophae and Elaeagnus root nodules [J]. J Anhui Agric Sci, 2010, 38(11): 33 − 36. [37] 张小民, 王岚, 林美珍, 等. 论沙棘根系与功能Ⅱ——Frankia菌侵染和结瘤[J]. 沙棘, 2006, 19(1): 4 − 14. ZHANG Xiaomin, WANG Lan, LIN Meizhen, et al. On the root system and function of seabuckthorn Ⅱ: Frankia infection and nodulation [J]. Seabuckthorn Hippophae, 2006, 19(1): 4 − 14. [38] FINKEL O M, SALAS-GONZÁLEZ I, CASTRILLO G, et al. A single bacterial genus maintains root growth in a complex microbiome [J]. Nature, 2020, 587: 103 − 108. [39] TRUJILLO M E, RIESCO R, BENITO P, et al. Endophytic actinobacteria and the interaction of Micromonospora and nitrogen fixing plants[J/OL]. Front Microbiol, 2015, 6: 1341[2021-01-12]. doi: 10.3389/fmicb.2015.01341. [40] CARRO L, PUJIC P, TRUJILLO M E, et al. Micromonospora is a normal occupant of actinorhizal nodules [J]. J Biosci, 2013, 38(4): 685 − 693. [41] HOCHER V, ALLOISIO N, BOGUSZ D, et al. Early signaling in actinorhizal symbioses [J]. Plant Signaling Behav, 2011, 6(9): 1377 − 1379. -
-
链接本文:
https://zlxb.zafu.edu.cn/article/doi/10.11833/j.issn.2095-0756.20210246
 点击查看大图
点击查看大图
计量
- 文章访问数: 2266
- HTML全文浏览量: 523
- PDF下载量: 48
- 被引次数: 0
