





-
中药荆芥是唇形科Labiatae植物荆芥Schizonepeta tenuifolia干燥后的地上部分,有解表祛风、透疹止血等功效[1]。荆芥的挥发油、黄酮等活性成分被广泛用于医药、食品和化工等领域[2-3]。
HD-Zip (Homeodomain-leucine zipper protein)基因家族是植物界一类特有的转录因子,在植物的生长发育、适应环境及胁迫应答等方面起到重要作用。HD (Homeodomain)蛋白是由Homeobox (HB)基因编码的高度保守的蛋白质结构域,由60个氨基酸组成。该蛋白中存在1个特征性的三螺旋结构,可以特异结合DNA序列,以此对基因进行调控[4−5]。此外,HD-Zip基因家族还有1个亮氨酸拉链保守结构域(leucine zipper-loop-zipper,LZ),这是蛋白形成二聚体所必需的结构。根据蛋白的序列保守性、蛋白功能、基因结构等,将该家族分为4个亚家族:HD-Zip Ⅰ ~Ⅳ[6]。Ⅰ亚家族主要参与非生物胁迫及环境适应性;Ⅱ亚家族主要与生长素响应相关;Ⅲ亚家族主要参与不同的发育事件,例如顶端分生组织、维管束的发育,还与植物激素调控相关;Ⅳ亚家族主要在植物的表皮中特异性表达,主要调节表皮的分化、毛状体形成等[7]。
目前,HD-Zip基因家族在多种植物中被鉴定并表征,例如拟南芥Arabidopsis thaliana[8]、水稻Oryza sativa[9]、小麦Triticum aestivum[10]等,但尚未有荆芥HD-Zip基因家族的相关研究。本研究以荆芥的基因组作为基础,利用生物信息学方法系统鉴定荆芥HD-Zip基因家族成员,并对其蛋白质理化性质、染色体定位、基因结构、共线性分析以及不同时期的表达规律进行分析,为今后深入研究荆芥基因家族的功能和调控机制奠定基础。
-
基于已知的HD-Zip基因家族的保守结构域,在荆芥基因组数据中进行初步筛选,利用TBtools (v1.98741)的“Blast Compare Two Seqs”,下载的蛋白序列为Query Seq,荆芥基因组的蛋白序列为Subject Seq,设置E-value为10−10进行比对[11]。根据HD-Zip基因家族在美国国家生物技术信息中心(NCBI)中的比对结果,得到目的基因编码蛋白的保守结构域,使用“Visualize NCBI CDD Domain Pattern”进行保守结构域的可视化。利用在线网站ExPASy (
https://www.expasy.org/ )对蛋白序列的基本理化性质,如氨基酸数目、等电点和分子质量等进行预测。 -
在NCBI在线网站上下载已被表征的HD-Zip基因家族的蛋白序列。将经过筛选的荆芥HD-Zip蛋白序列与下载的蛋白序列利用MEGA X进行最大似然 (ML)进化树的构建。选择最优氨基酸替代模型,根据氨基酸模型结果构建ML树,设置bootstrap为1000,partial deletion为80%。利用在线网站iTOL (
https://itol.embl.de/# )对进化树进行美化。 -
在荆芥基因组中搜索HD-Zip基因在染色体上的具体位置和每条染色体的总长度,利用TBtools中的“Visualize Gene Structure (Basic)”功能,对筛选的基因ID进行基因结构的可视化。利用在线网站MEME (
https://meme-suite.org/meme/tools/meme )对筛选的荆芥HD-Zip基因编码的蛋白序列进行蛋白保守基序预测,设置基序数量为10个,选择“Zero or One Occurence Per Sequence (zoops)”分布基序。采用TBtools中的“Visualize MEME/MSAT Motif Pattern”进行保守基序的可视化处理。 -
利用TBtools的“Gene Location Visualize from GTF/GFF”进行基因在染色体分布的可视化。将筛选的基因序列利用TBtools中的“Gene Location Visualize fron GTF/GFF”功能进行染色体定位分析;提取荆芥HD-Zip基因序列的启动子部分(5′UTR上游2000 bp),利用PlantCARE在线网站(
http://bioinformatics.psb.ugent.be/webtools/plantcare/html/ )预测顺式元件并整理结果,再利用TBtools中的“Gene Structure View (Advanced)”对其进行可视化处理。使用MCScanX软件进行基因组内荆芥HD-Zip基因的共线性分析以及与拟南芥基因组间的共线性分析,并利用Circos软件绘制基因组内和基因组之间的共线性图谱。 -
根据HD-Zip基因ID于不同时期荆芥叶片(10、20、35 d)及根(35 d)的转录组数据中进行搜索,得到基因的FPKM (fragments per kilobase per million)值,利用TBtools的“HeatMap”绘制基因表达热图,探究HD-Zip基因家族的表达模式。
-
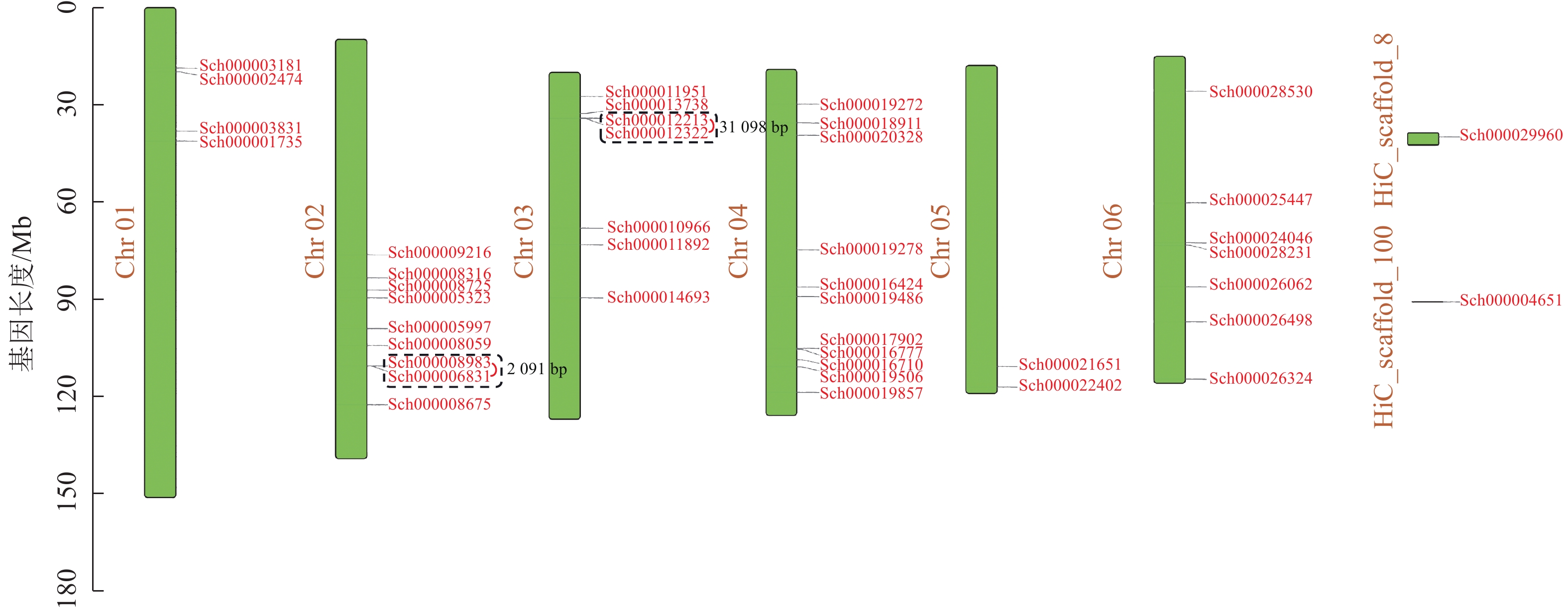
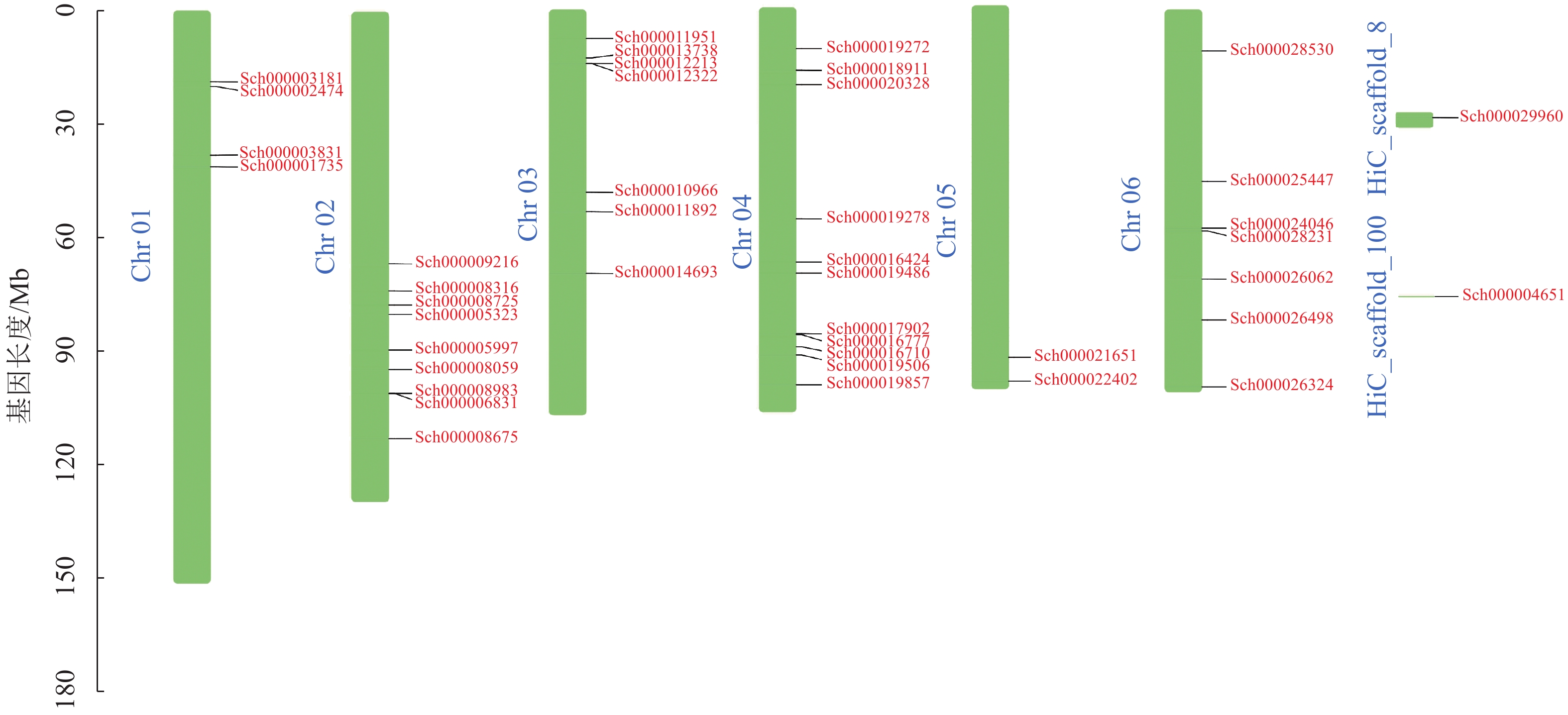
荆芥基因组大小为798 Mb,Q20(碱基被测错的概率为1%)为94.67%,Q30(碱基被测错的概率为1‰)为89.41%,说明测序质量较好(Q20≥93%、Q30≥86%),GC含量为39.34%,经过Hi-C组装后,共有696 Mb的基因组序列被定位到6条染色体上(Chr 01~06),占比91.38%。以上数据说明荆芥的基因组质量较好,有助于完整地挖掘HD-Zip基因家族。为了鉴定荆芥中HD-Zip基因,根据4个亚家族HD-Zip Ⅰ、Ⅱ、Ⅲ、Ⅳ的蛋白保守结构域进行筛选,共筛选到42条可能的HD-Zip基因家族序列,其中HD-Zip Ⅰ亚家族16条,HD-Zip Ⅱ亚家族7条,HD-Zip Ⅲ亚家族5条,HD-Zip Ⅳ亚家族14条,并通过在线网站Expasy网站进行蛋白分子量和等电点的预测(表1)。其中40条基因全部定位到对应染色体(Chr 01~06),Sch000029960和Sch000004651未锚定在染色体上(图1)。荆芥HD-Zip基因仅在2~4号染色体上集中分布,说明该基因家族在染色体上分布不均匀。荆芥HD-Zip的基因长度为528~2586 bp;分子量为20.33~94.18 kDa;等电点为4.59~9.05。因此,HD-Zip的基因和蛋白长度跨度较大,HD-Zip Ⅲ和Ⅳ的基因长度约2000 bp,HD-Zip Ⅰ和Ⅱ在1000 bp以下,该结果与分子量具有相关性,而等电点主要取决于氨基酸中酸性氨基酸和碱性氨基酸的数量比,大多数蛋白(76.2%)等电点小于7.0,证明荆芥HD-Zip可能是一类酸性蛋白。
表 1 荆芥HD-Zip基因家族的蛋白特征
Table 1. Protein characteristics of HD-Zip gene family in S. tenuifolia
亚家族 基因ID CDS长
度/bp蛋白长
度/个分子量/
kDa等电点 染色体 亚家族 基因ID CDS长
度/bp蛋白长
度/个分子量/
kDa等电点 染色体 HD-Zip Ⅰ Sch000003181 900 299 33.847 4.88 Chr 01 Sch000003831 528 175 20.331 8.58 Chr 01 Sch000016777 897 298 34.379 6.55 Chr 04 Sch000019486 822 273 31.100 4.59 Chr 04 HD-Zip Ⅲ Sch000001735 2562 853 93.306 5.74 Chr 01 Sch000026498 927 308 35.350 5.01 Chr 06 Sch000019857 2511 836 91.461 5.94 Chr 04 Sch000008725 822 273 31.034 4.83 Chr 02 Sch000014693 2493 830 91.021 5.84 Chr 03 Sch000005997 912 303 34.369 4.97 Chr 02 Sch000026324 2586 861 94.183 6.14 Chr 06 Sch000020328 966 321 35.730 4.81 Chr 04 Sch000028231 2529 842 92.434 6.17 Chr 06 Sch000016424 867 288 32.633 6.32 Chr 04 Sch000017902 879 292 33.207 6.07 Chr 04 HD-Zip Ⅳ Sch000026062 2166 721 79.071 5.98 Chr 06 Sch000002474 876 291 32.531 5.7 Chr 01 Sch000008059 2163 720 79.056 6.23 Chr 02 Sch000008983 696 231 26.832 6.32 Chr 02 Sch000029960 2181 726 79.526 5.64 HiC_scaffold_8 Sch000006831 693 230 26.615 6.96 Chr 02 Sch000011892 2418 805 87.913 5.79 Chr 03 Sch000019278 600 199 22.673 8.44 Chr 04 Sch000009216 2196 731 79.888 5.79 Chr 02 Sch000021651 546 181 21.788 5.84 Chr 05 Sch000019506 2400 799 87.073 6.04 Chr 04 Sch000013738 654 217 24.934 7.59 Chr 03 Sch000012322 2490 829 91.502 5.41 Chr 03 Sch000016710 621 206 24.472 5.44 Chr 04 Sch000012213 2490 829 91.502 5.41 Chr 03 Sch000004651 2490 829 91.502 5.41 HiC_scaffold_100 HD-Zip Ⅱ Sch000028530 891 296 327.360 7.52 Chr 06 Sch000018911 2070 689 77.061 6.28 Chr 04 Sch000010966 879 292 32.623 7.62 Chr 03 Sch000005323 1944 647 73.264 7.27 Chr 02 Sch000019272 795 264 294.950 8.59 Chr 04 Sch000022402 2307 768 84.180 5.81 Chr 05 Sch000008316 783 260 29.093 8.12 Chr 02 Sch000024046 2274 757 84.279 6.15 Chr 06 Sch000025447 879 292 32.351 9.05 Chr 06 Sch000008675 1884 627 69.905 6.18 Chr 02 Sch000011951 645 214 24.334 8.24 Chr 03 说明:CDS指蛋白编码区 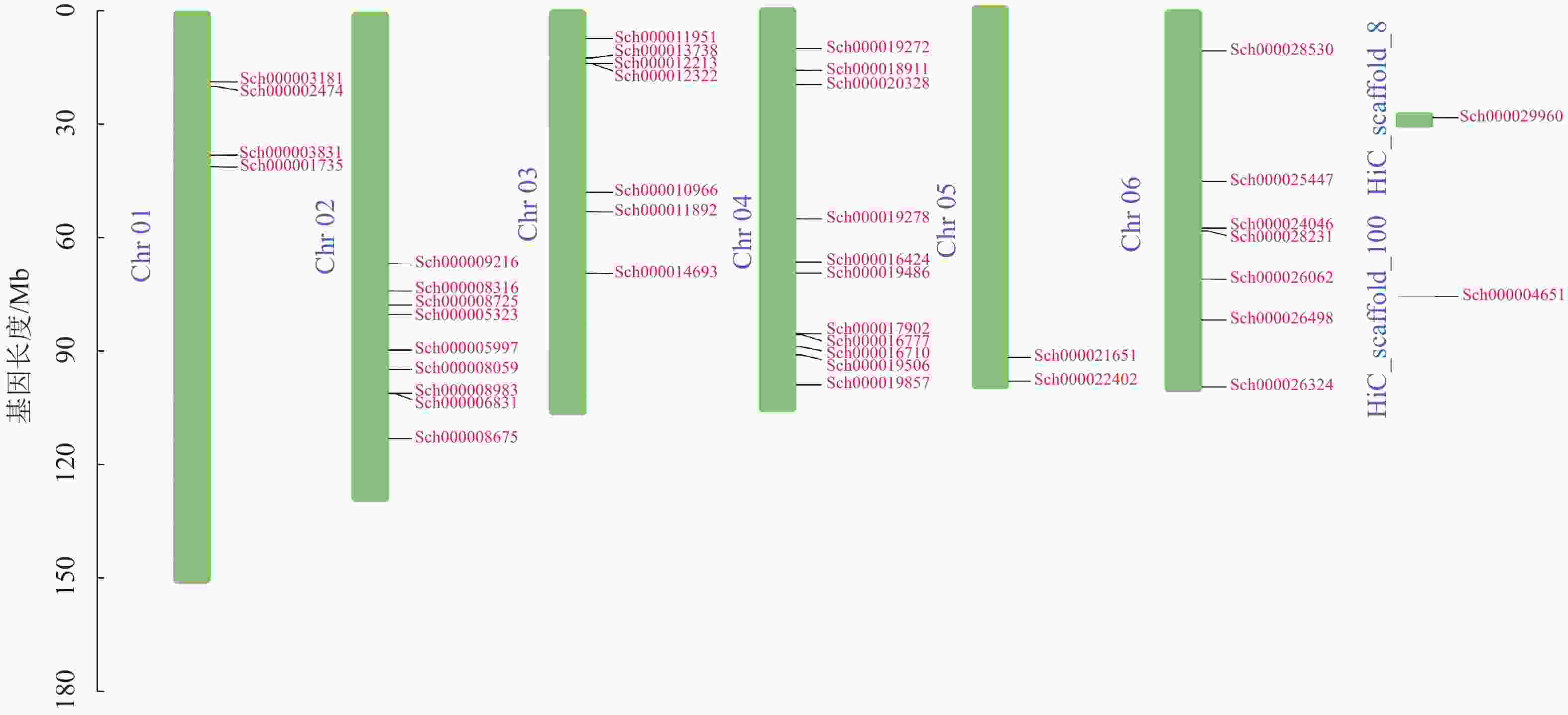
图 1 荆芥HD-Zip基因家族的染色体定位
Figure 1. Chromosome mapping of HD-Zip gene family in S. tenuifolia
-
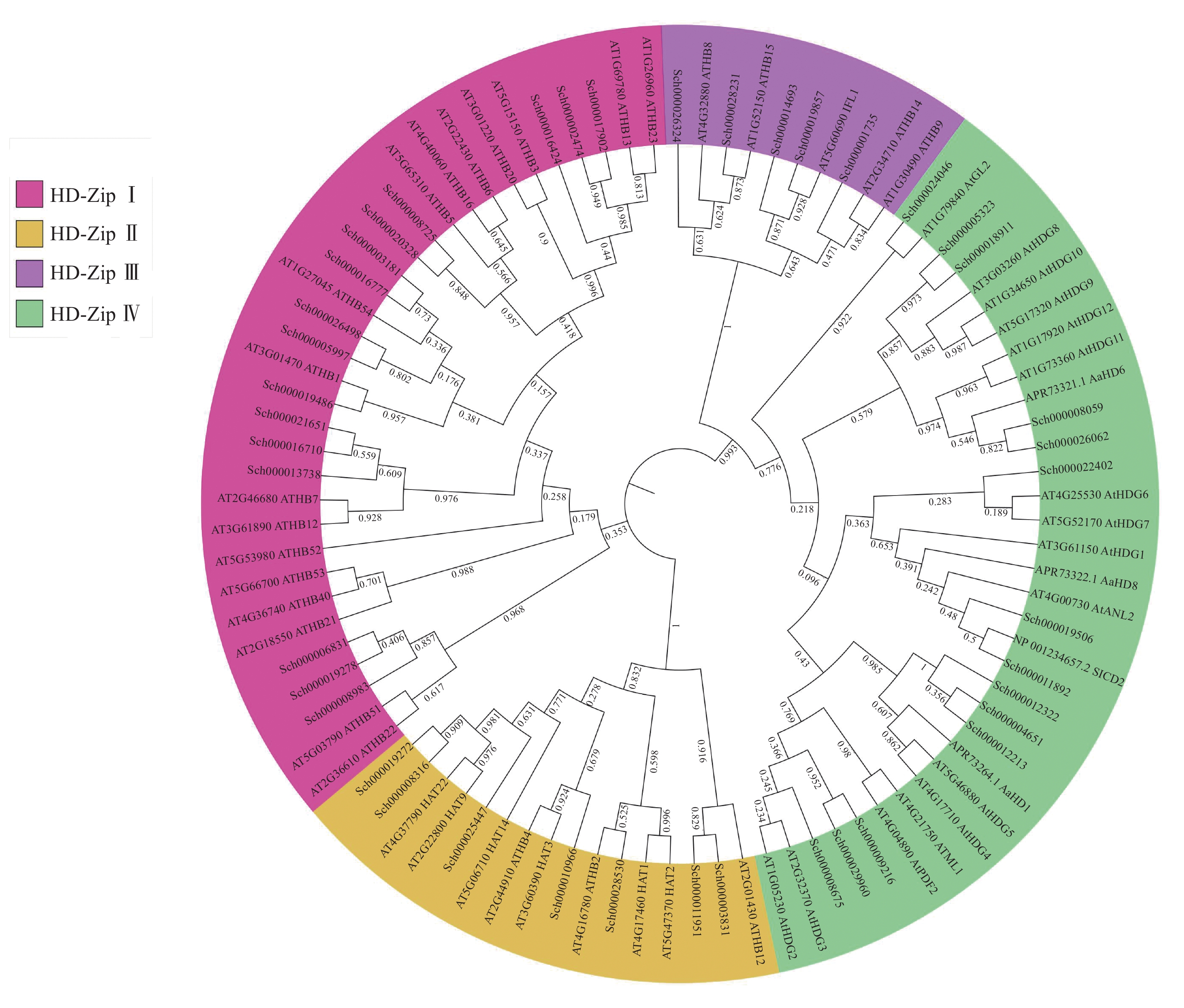
将以上42条蛋白序列与已知的HD-Zip蛋白序列进行ML树的构建(图2),可知:荆芥的HD-Zip和拟南芥及其他物种HD-Zip的蛋白序列被聚为四大支,与已表征HD-Zip基因家族的4个亚家族分类一致,且在荆芥基因组中,每个亚家族基因的占比与拟南芥的HD-Zip Ⅰ ~Ⅳ之间的比例相似,其中HD-Zip Ⅰ与Ⅳ占比最大,HD-Zip Ⅲ占比最少。从进化树中可以发现:HD-Zip Ⅲ先与Ⅳ聚为一支,再与HD-Zip Ⅰ和Ⅱ聚为一支,说明HD-Zip Ⅲ可能与Ⅳ的亲缘关系更近。
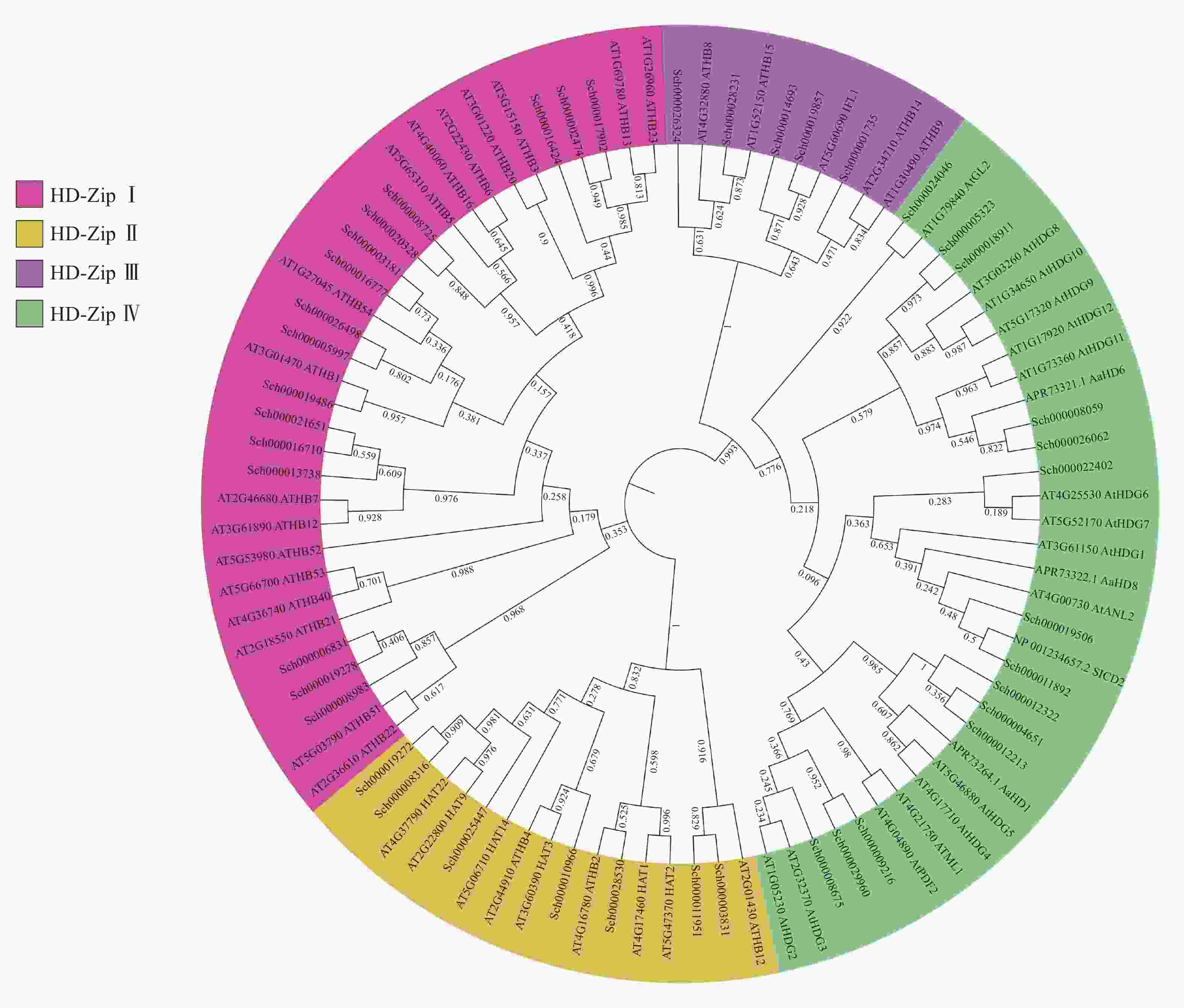
图 2 荆芥与拟南芥及其他物种HD-Zip基因家族的最大似然值进化树
Figure 2. ML evolutionary tree of HD-Zip gene family between S. tenuifolia and A. thaliana and other species
-
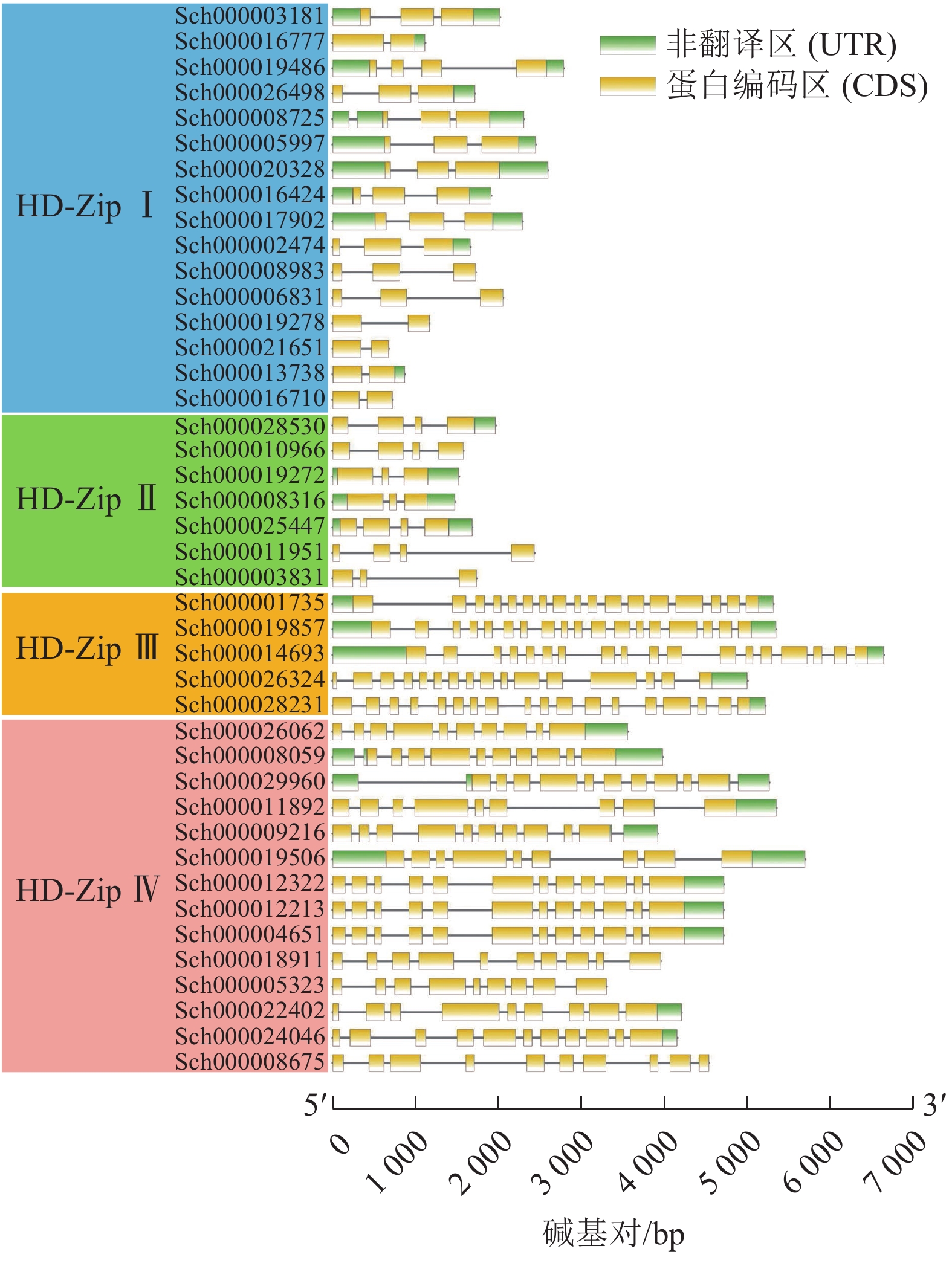
利用TBtools软件绘制荆芥HD-Zip基因结构图,分析基因内含子和外显子的分布情况。图3显示:HD-Zip Ⅰ与Ⅱ的基因长度较为相近,内含子1~3个(实线),外显子2~4个(黄色标识),基因结构比较简单。HD-Zip Ⅲ与Ⅳ基因长度较为接近,内含子8~17个,外显子9~17,其中HD-Zip Ⅲ的内含子和外显子的数量最多。以上基因结构和长度结果与ML进化树聚类结果较为一致。
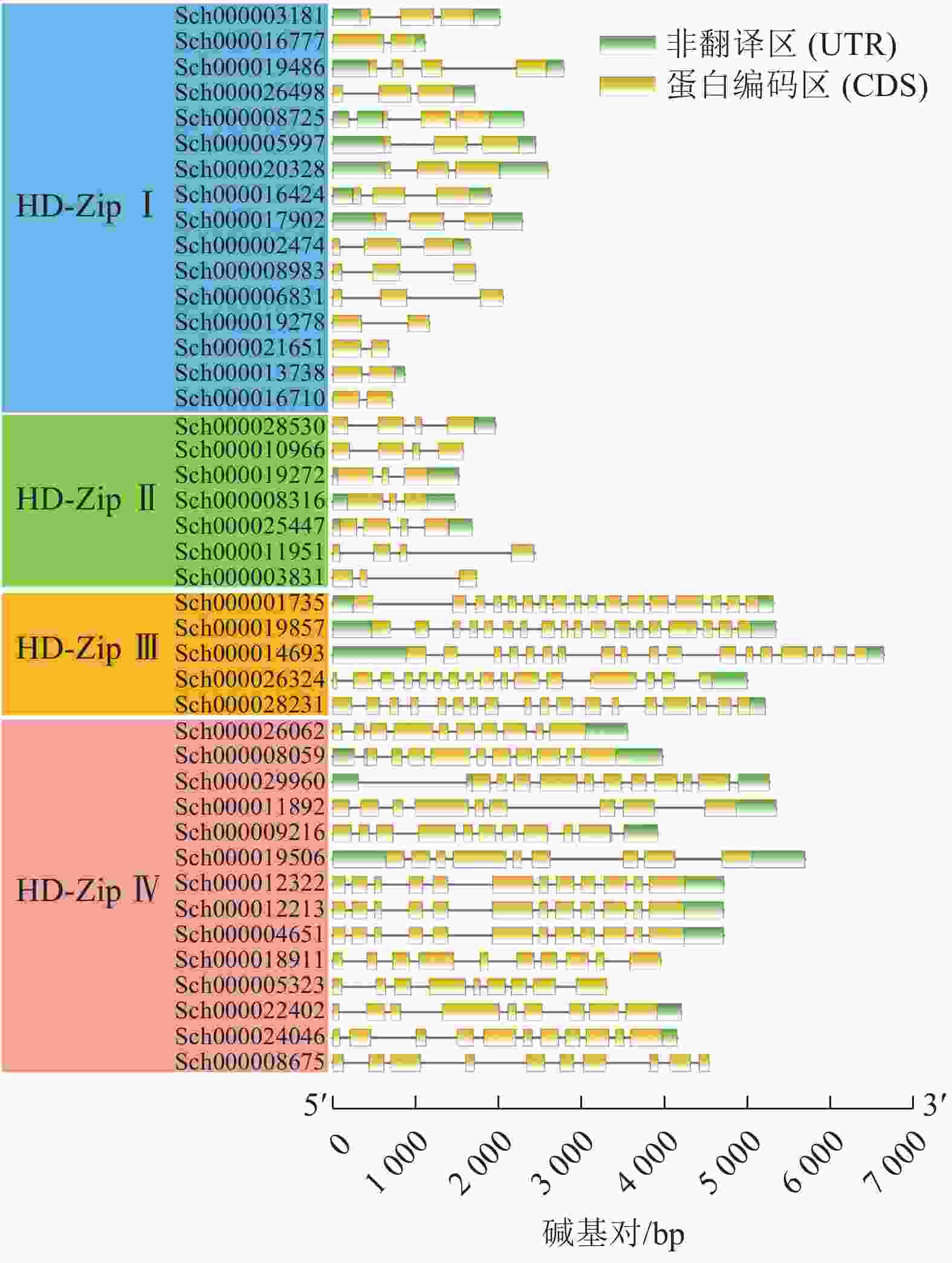
图 3 荆芥HD-Zip基因家族的基因结构分析
Figure 3. Gene structure analysis of HD-Zip gene family in S. tenuifolia
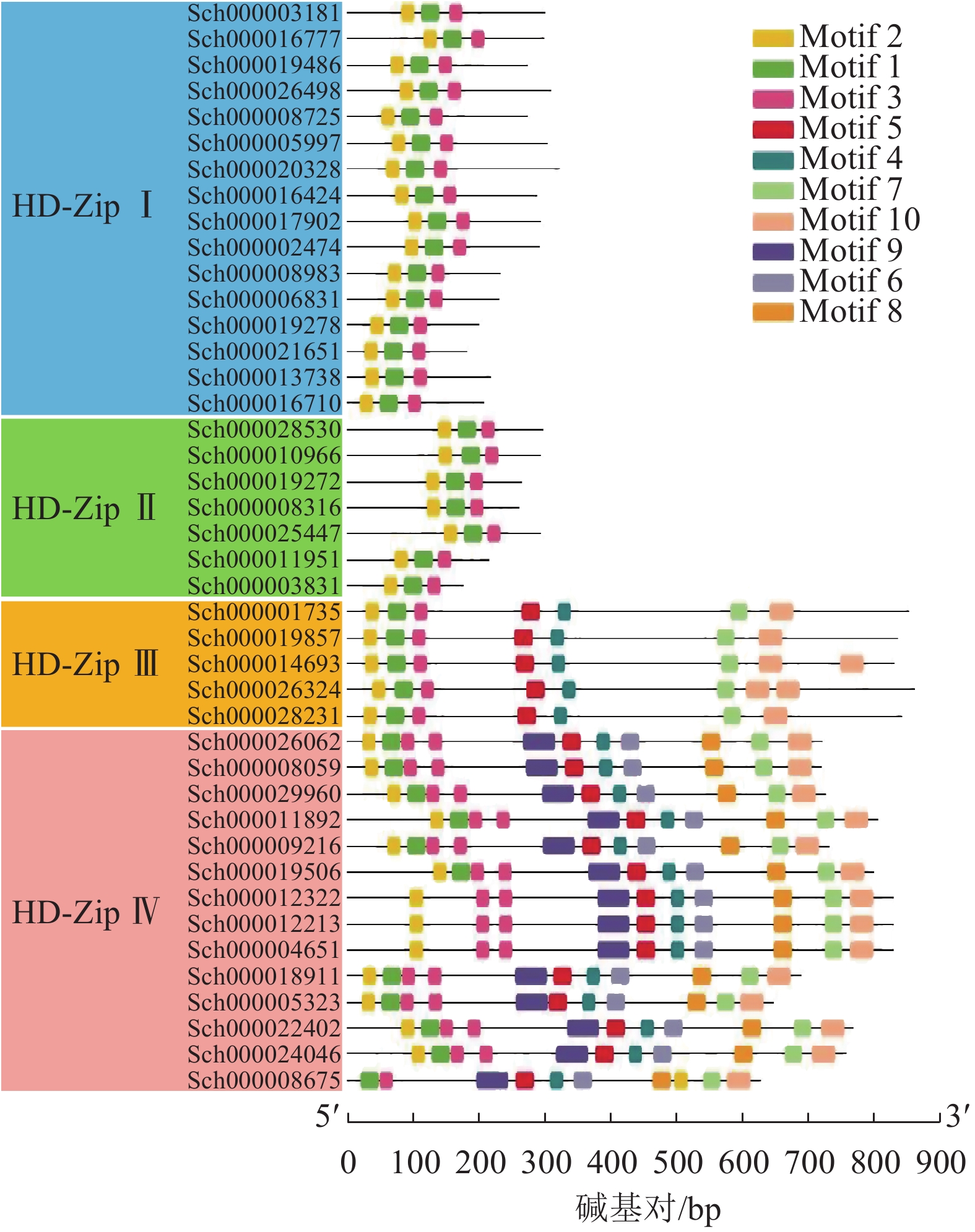
利用在线网站MEME对42条HD-Zip基因家族的蛋白序列进行保守基序(Motif)的检索,一共确认了10个不同的基序(图4)。其中,所有蛋白均存在Motif 1~3,这3个保守基序构成了HD-Zip基因家族特征的保守基序HD、LZ。HD-Zip Ⅲ和Ⅳ的Motif 4、Motif 5构成HD-Zip Ⅲ和Ⅳ特有的START保守结构域。从Motif结构分布上看到,HD-ZipⅢ和Ⅳ的Motif最为丰富,可能具有多样的生物学功能,每个亚家族之间的Motif分布较为一致。

图 4 荆芥HD-Zip基因家族的保守基序分析
Figure 4. Conservative motif analysis of HD-Zip gene family in S. tenuifolia
-
提取荆芥HD-Zip的5′UTR上游的2 kb序列为启动子序列,利用在线网站PlantCARE进行顺式元件的预测,其中光响应的顺式元件出现频率最高,其次为脱落酸响应元件,MeJA响应元件,厌氧感应元件以及MYB结合的位点(图5)。说明该基因家族可能与以上的生物学功能相关。
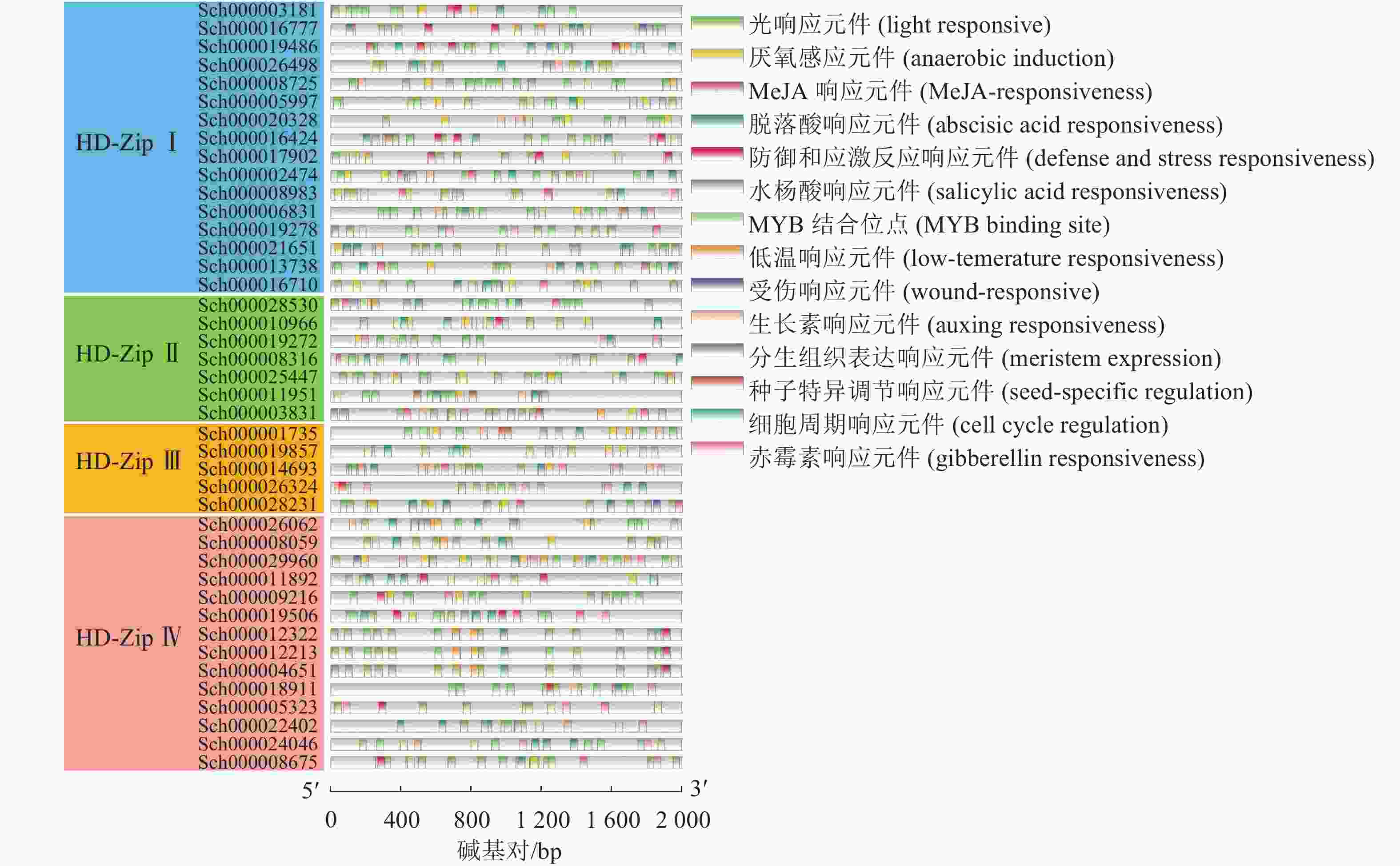
图 5 荆芥HD-Zip基因家族的顺式作用元件分布
Figure 5. Distribution of cis-acting elements of HD-Zip gene family in S. tenuifolia
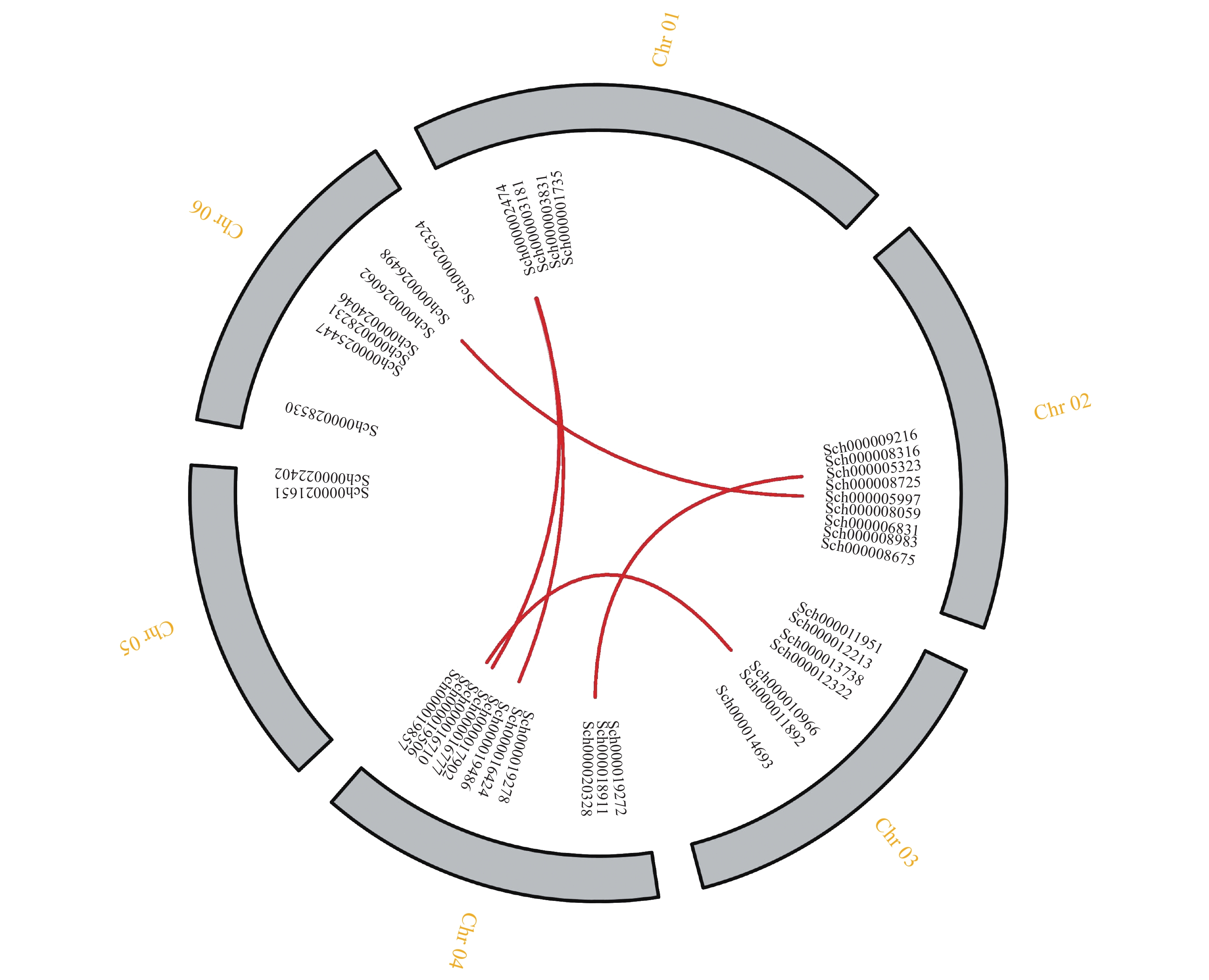
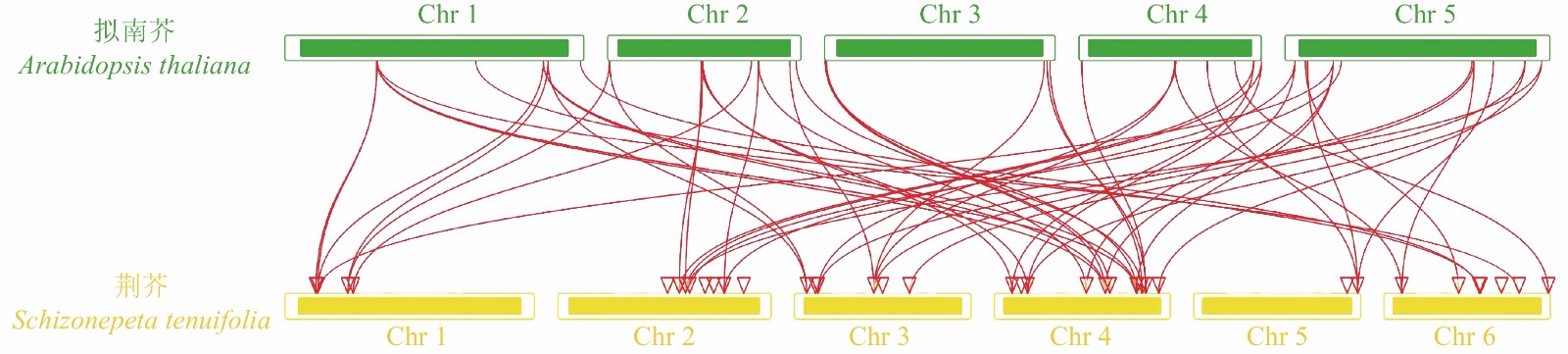
对荆芥的42个HD-Zip家族基因进行基因组内串联重复分析,发现Sch000008983和Sch000006831在Chr 02上串联重复,Sch000012213与Sch000012322在Chr 03上串联重复(图6);经过基因组内的共线性分析发现,荆芥的9个HD-Zip家族基因在基因组内存在共线性,说明成对的共线性基因可能具有极为相似的功能(图7)。通过荆芥与拟南芥的基因组之间的共线性分析发现:一共有37对共线性的HD-Zip基因(图8)。综上,通过与拟南芥HD-Zip基因构建进化树分析及共线性分析,有助于利用拟南芥的基因功能推断荆芥HD-Zip中相应基因的功能。
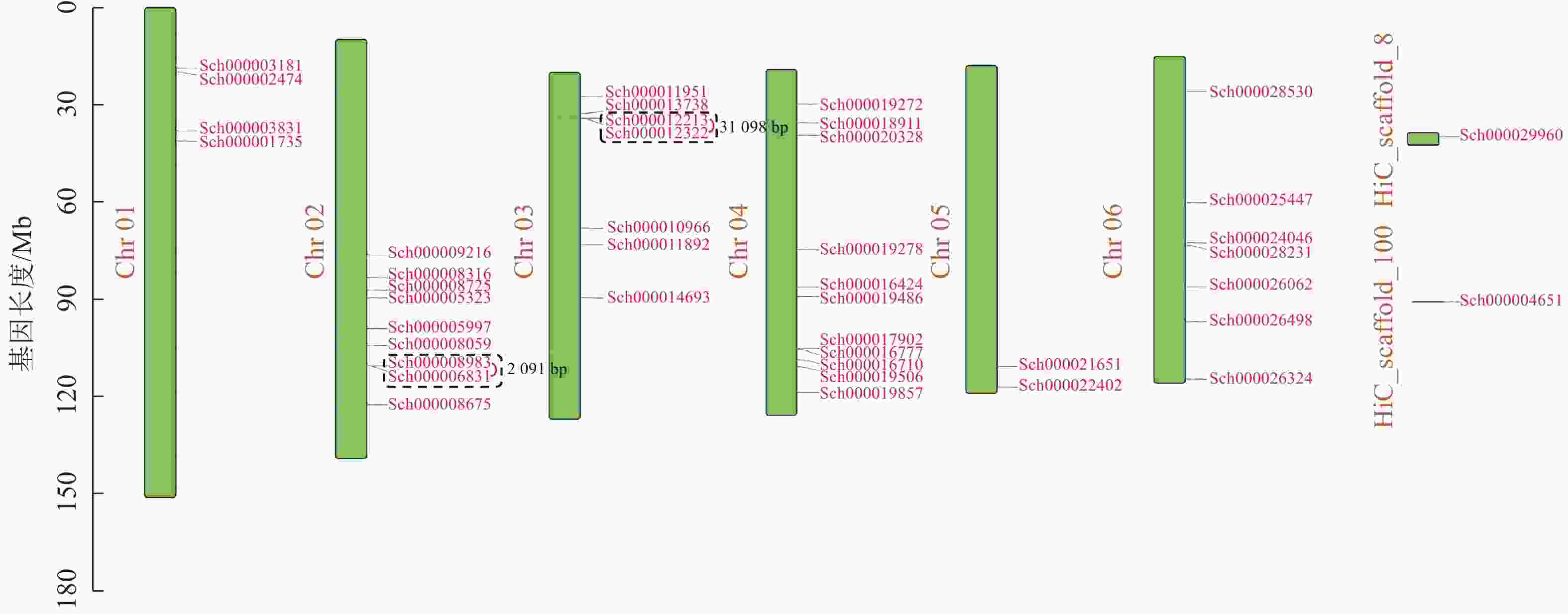
图 6 荆芥HD-Zip基因家族的组内串联重复分析
Figure 6. Tandem repeat analysis of HD-Zip gene family in genome of S. tenuifolia
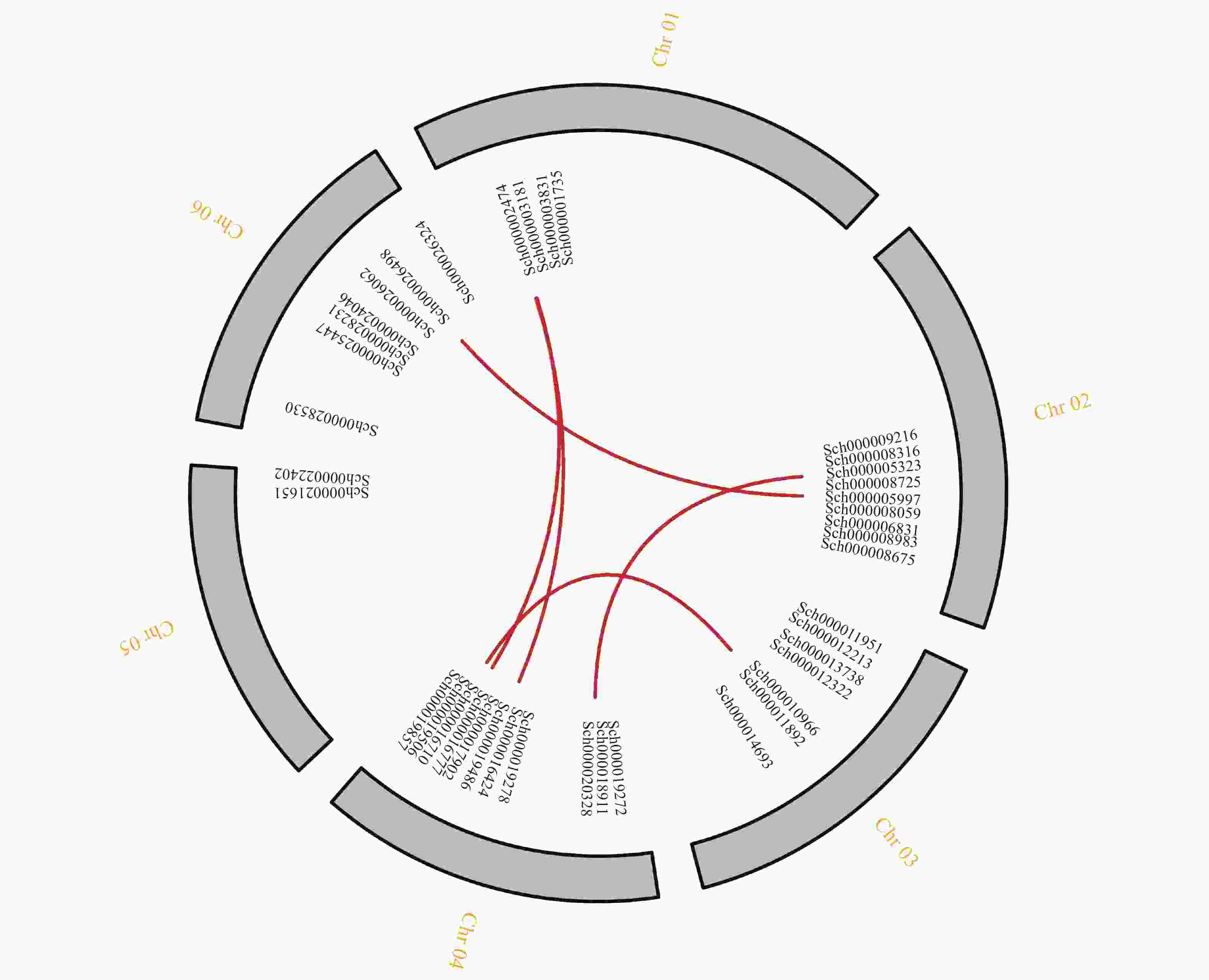
图 7 荆芥HD-Zip基因家族的组内共线性分析
Figure 7. Intra-group collinearity analysis of HD-Zip gene family in S. tenuifolia
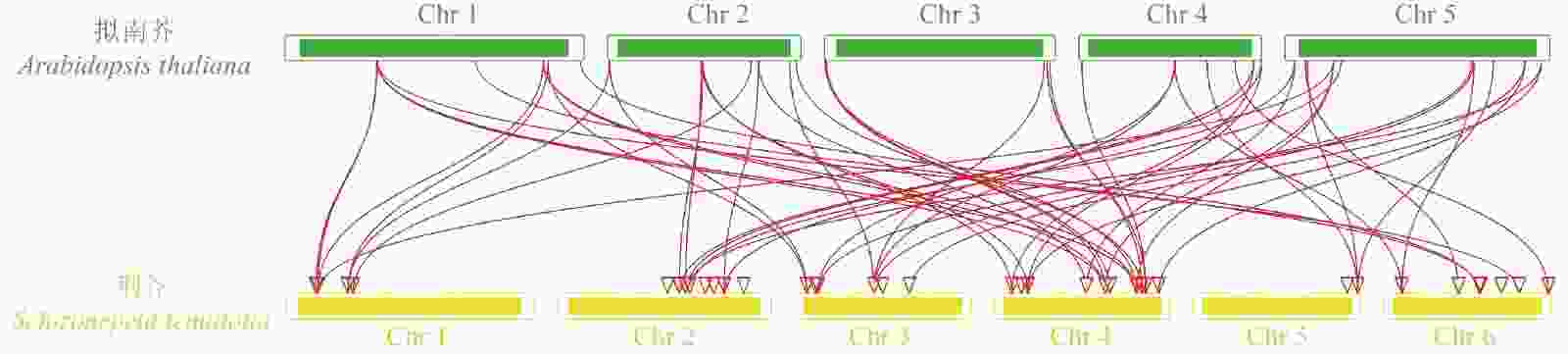
图 8 荆芥HD-Zip与拟南芥基因组之间的共线性分析
Figure 8. Collinear analysis of HD-Zip gene between S. tenuifolia and A. thaliana genomes
-
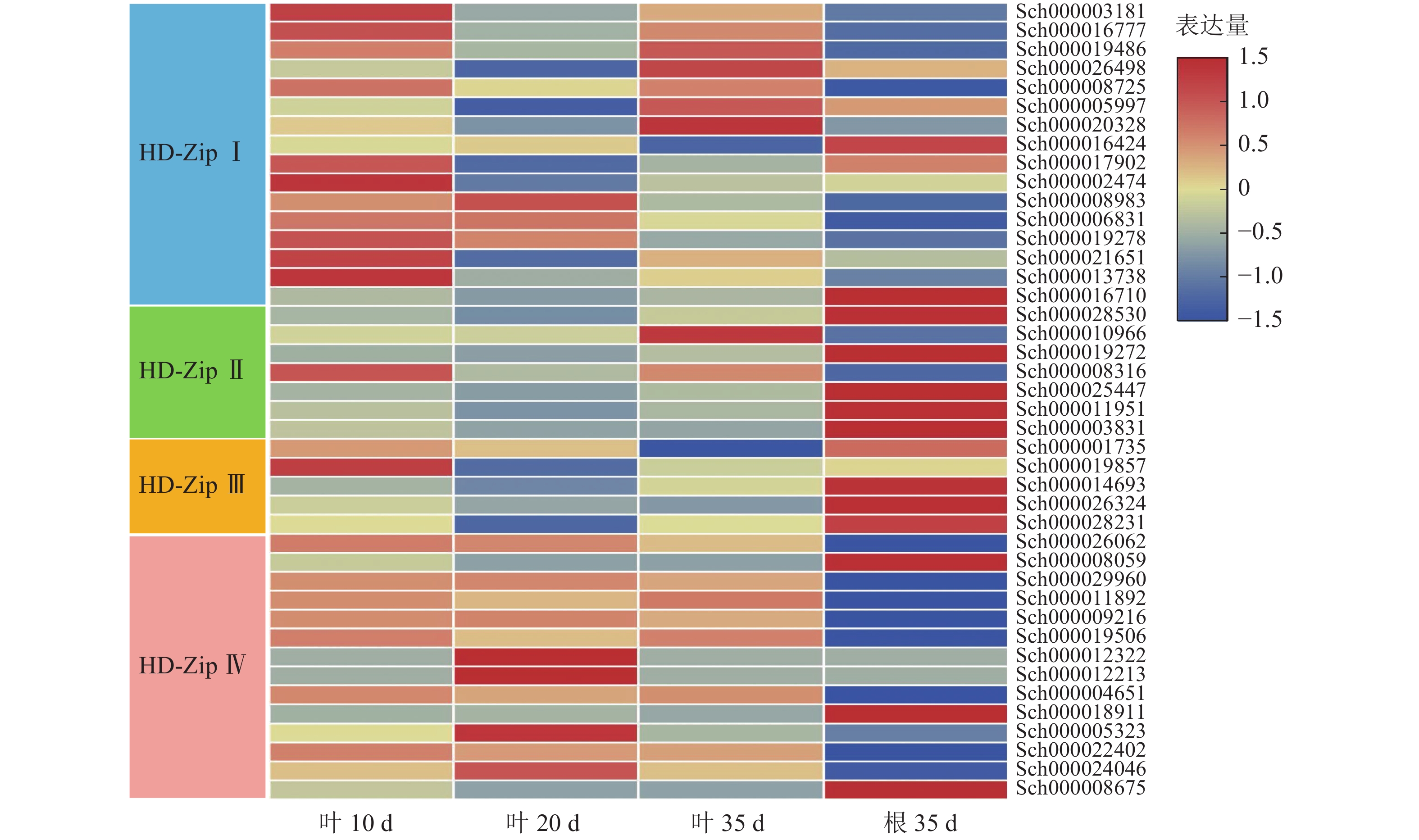
根据课题组前期观察,10 d幼苗的叶子和茎具有丰富的指状腺毛,20 d幼苗的叶子和茎具有较多的头状腺毛和腺鳞,35 d幼苗的叶子和茎具有丰富的腺鳞。因此,对荆芥不同生长时期叶片(10、20、35 d)及根(35 d)进行转录组分析,发现HD-Zip Ⅰ主要在幼叶10 d中表达,HD-ZipⅡ和Ⅲ主要在根中表达,HD-Zip Ⅳ亚家族主要在叶中表达(图9)。研究发现:HD-Zip Ⅳ基因主要调节表皮的分化[12],结合荆芥腺毛的分布情况,推测荆芥的HD-Zip Ⅳ与荆芥腺毛和非腺毛的形成与分化相关。
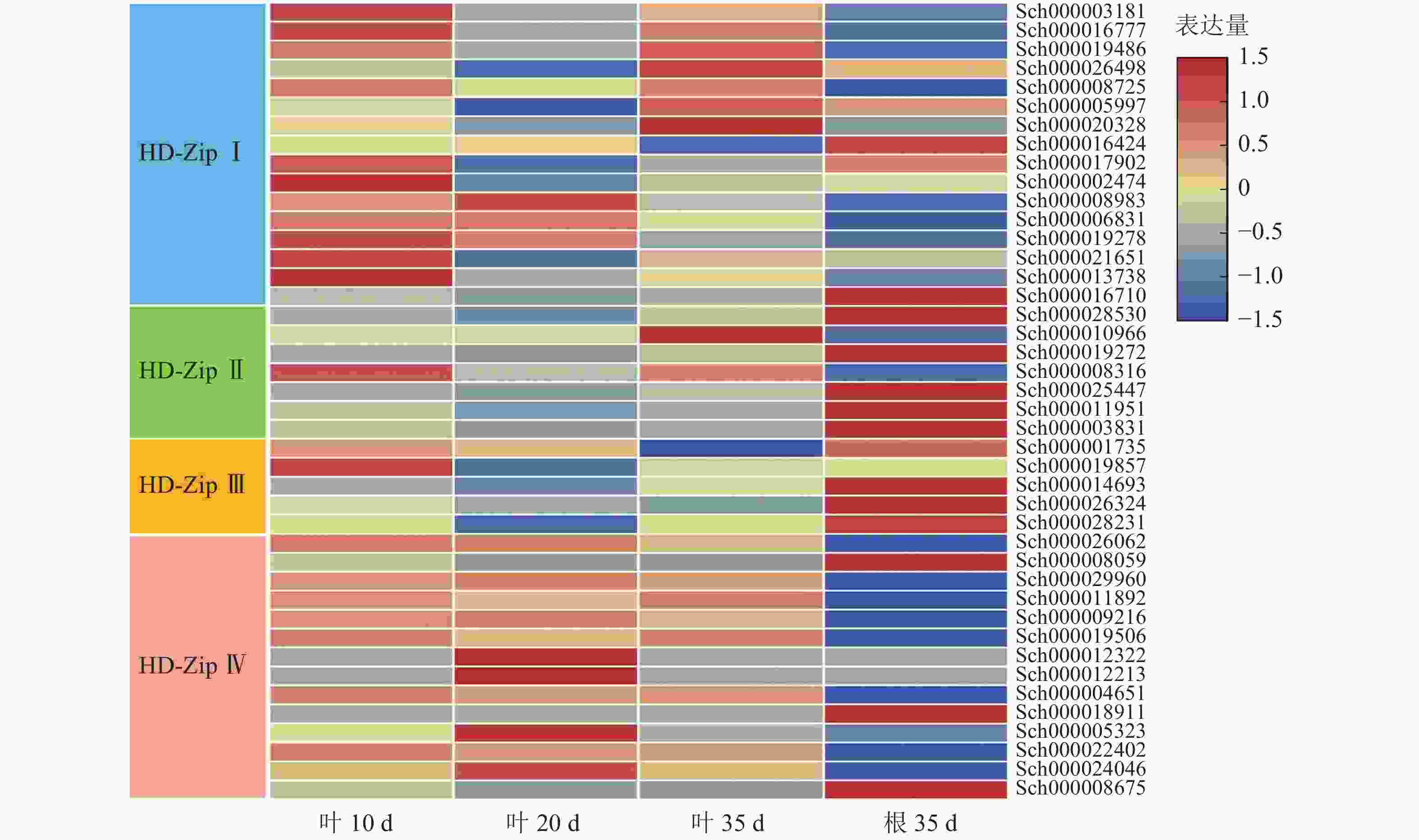
图 9 HD-Zip家族基因表达模式
Figure 9. HD-Zip family gene expression pattern
-
本研究从全基因组水平对荆芥的HD-Zip基因家族进行了系统的研究,共鉴定到42个HD-Zip家族的基因,根据识别的DNA序列、结构域、蛋白功能,可将这些序列分为4个亚家族,分别为HD-Zip Ⅰ ~Ⅳ,这与拟南芥、小麦、水稻、玉米Zea mays、土豆Solanum tuberosum、烟草Nicotiana tabacum等中的分类一致[7-9, 13-15]。HD-Zip Ⅰ只含有高保守的HD结构域和位于HD结构域羧基端的LZ结构域;HD-Zip Ⅱ除了HD-Zip Ⅰ具有的HD和LZ保守结构域外,还存在1个高度保守的N-末端;HD-Zip Ⅲ具有HD和LZ保守结构域,以及类固醇合成急性调节蛋白相关的脂质转运结构域(START)和氨基酸序列羧基端的MEKHLA基序,其中START结构域的长度为220个氨基酸且可以结合并转移脂质,MEKHLA基序与许多非生物胁迫应答相关[16-17];HD-Zip Ⅳ结构与HD-Zip Ⅲ非常相似,具有HD、LZ、START结构域,但缺失了MEKHLA基序[18]。荆芥的HD-Zip Ⅰ和Ⅳ亚家族的基因所占比例最高,这与拟南芥HD-ZipⅠ和Ⅳ的比例相似。基因的进化树结果显示:HD-Zip Ⅰ与Ⅱ亲缘关系更近,HD-Zip Ⅲ与Ⅳ亲缘关系更近,由此可以推测以上2个分支可能是由相同的祖先进化而来,或者Ⅳ是由Ⅲ进化来,但在分化过程中丢失了MEKHLA基序[19]。结合基因的结构来看,HD-Zip Ⅲ和Ⅳ的结构比HD-Zip Ⅰ与Ⅱ的结构更为复杂,以上结果说明可能HD-Zip Ⅲ与Ⅳ相比于HD-ZipⅠ和Ⅱ进化程度更高,基因结构更为复杂,以上结果与保守结构域分析和进化树的分析结果一致。这说明HD-Zip家族在物种的亚群内部较为保守,但其具体的基因功能可能会由于基因复制或者进化,以及物种间的差异性从而出现一定的差异。
分析启动子发现:在每个亚族内部的基因启动子区顺式作用元件类型基本相同,例如MYB结合位点、脱落酸响应元件以及MeJA响应元件在HD-Zip Ⅳ高频出现。同时,同一亚族基因编码蛋白的保守基序也基本相同,HD-Zip Ⅰ ~Ⅳ的表达分析发现:HD-Zip Ⅰ ~Ⅳ具有不同的表达偏好性,说明荆芥中不同HD-Zip家族不同亚家族可能具有不同的生物学功能,但同一亚族各基因的生物学功能基本相同。
有研究表明:HD-Zip Ⅳ在表皮中特异表达,参与植物表皮细胞的分化,调节毛状体(腺毛和非腺毛)等形成与发育。如烟草中的NtHDG2,拟南芥的PDF2,黄花蒿Artemisia annua的AaHD1和AaHD8,番茄Solanum lycopersicum的SlCD2和SlWo均对毛状体具有调控作用,属于HD-Zip Ⅳ[14, 20-22]。本研究中发现荆芥的HD-Zip Ⅳ亚家族基因大部分在叶片表达,推测可能这些基因与毛状体的发育相关。结合拟南芥与荆芥HD-Zip基因家族的共线性分析,可以推测荆芥HD-Zip基因家族的生物学功能。结合文献,发现Sch000029960与AT4G21750.1及AT4G04890.2为同源基因,AT4G21750.1及AT4G04890.2分别编码拟南芥的GL2-like和PDF2,与拟南芥的表皮发育密切相关。Sch000024046与AT1G79840.2为同源基因,AT1G79840.2编码GL2,在拟南芥中影响表皮细胞的特性,包括毛状体、根毛发育等[23]。在荆芥的叶、茎、花穗等多个部位表面分布着多种腺毛及非腺毛,其中,盾状腺毛即腺鳞被认为是荆芥产生挥发油的“品质载体”[24-25],但是调控荆芥腺鳞生长发育的分子机制还未被报道,本研究中筛选的HD-Zip Ⅳ亚基因家族可能为腺鳞发育调控的候选基因。通过对候选基因功能的验证、共表达分析等为腺鳞生长发育分子机制的阐明提供线索,同时为提高荆芥药用品质提供理论基础。
-
本研究在荆芥全基因水平上筛选到42条HD-Zip基因序列,并对以上序列的基因结构、保守基序、顺式作用元件等进行了分析。系统发育分析可将42条序列分为4个亚家族(HD-Zip Ⅰ ~Ⅳ)。通过与拟南芥基因组之间的共线性分析、表达模式分析等推测,荆芥的HD-Zip Ⅳ亚家族基因可能在毛状体发育过程中起到重要作用。这些结果为后续荆芥的HD-Zip基因家族的功能研究及表征提供了理论基础。
Genome-wide identification and expression analysis of HD-Zip gene family in Schizonepeta tenuifolia
-
摘要:
目的 鉴定荆芥Schizonepeta tenuifolia的HD-Zip基因家族,利用生物信息学方法分析其在全基因组中的分布和相关特征以及在不同时期中的表达规律,为该家族基因的进一步研究奠定基础。 方法 根据已经表征的HD-Zip基因,筛选荆芥基因组内的HD-Zip基因序列,利用MEME、PlantCARE、NCBI、MEGA X、MCScanX、Circos等在线网站及软件对蛋白序列进行基本理化性质分析、进化树构建、染色体定位、基因结构分析、共线性基因分析等。 结果 在荆芥全基因组中共鉴定到42条HD-Zip基因序列,它们可被分为4个亚家族,分别含有16、7、5、14个基因,亚家族之间的基因长度、结构及保守基序差异显著,但在亚家族内部保守,荆芥基因组与拟南芥Arabidopsis thaliana基因组共线性分析发现有37对基因,可能具有相似的生物学功能。荆芥的4个亚家族基因的顺式元件中均高频出现了光响应、脱落酸响应、MeJA响应等元件,在不同生长时期的叶片及部位的转录组数据中具有不同的表达趋势,Ⅰ亚家族主要在幼叶中表达,Ⅱ和Ⅲ亚家族主要在根中在表达,Ⅳ亚家族主要在叶中表达。 结论 在荆芥基因组中共获得42条HD-Zip基因序列,被分为4个亚家族(HD-ZipⅠ~Ⅳ),亚家族内部高度保守,亚家族之间差异显著,其基因结构、保守结构域及表达模式不同。亚家族Ⅰ和Ⅱ,亚家族Ⅲ和Ⅳ亲缘关系更近,HD-Zip基因具有组织表达差异性,协同调控了荆芥的生长发育和次生代谢。图9表1参25 -
关键词:
- 荆芥 /
- HD-Zip基因家族 /
- 表达分析 /
- 系统进化
Abstract:Objective This study, with the identification of the HD-Zip gene family in Schizonepeta tenuifolia and an bioinformatic analysis of its distribution and related characteristics in the whole genome and expression pattern in different stages, is aimed to provide a basis for further study of this gene family. Method Genes in the genome were first screened in accordance with the characterized HD-Zip genes before MEME, Plant CARE, NCBI, MEGA X, MCScanX and Circos were used for the analysis of the basic properties of protein sequences, the construction of Maximum Likeliood (ML) tree, mapping of chromosomes and the analysis of gene structure and colinear gene respectively. Result (1) A total of 42 HD-Zip genes were identified and they could be divided into four subgroups(Ⅰ−Ⅳ), containing 16, 7, 5, 14 genes respectively. (2) The gene length, structure and Motif of the subgroups varied significantly from each though relatively reserved with in each of them. (3) The collinear analysis of the genome of S. tenuifolia and Arabidopsis thaliana showed that 37 pairs of genes may have similar biological functions with light response, abscisic acid response and MeJA response found in cis-elements of 4 subfamily genes promoters of S. tenuifolia, showing different expression profiles in the transcriptome data of different growth stages of leaves and different parts. (4) The HD-ZipⅠ was mainly expressed in young leaves, HD-Zip Ⅱ and Ⅲ were mainly expressed in roots whereas HD-Zip Ⅳ was mainly expressed in leaves. Conclusion The total 42 HD-Zip genes obtained from S. tenuifolia genome were divided into 4 subgroups (Ⅰ−Ⅳ) with highly conserved genes and significant differences among subgroups with their gene structure, motif, and expression pattern being different. HD-ZipⅠ and Ⅱ, and HD-ZipⅢ and Ⅳ are more closely related to each other with HD-Zip genes expressed specifically indifferent tissues and synergistically regulating the growth, development and secondary metabolism of S. tenuifolia. The results of this study provide a bioinformatic reference for the further study of this family’s biological functions. [Ch, 9 fig. 1 tab. 25 ref.] -
Key words:
- Schizonepeta tenuifolia /
- HD-Zip gene family /
- expression analysis /
- system evolution
-
图 1 荆芥HD-Zip基因家族的染色体定位
Figure 1 Chromosome mapping of HD-Zip gene family in S. tenuifolia
图 2 荆芥与拟南芥及其他物种HD-Zip基因家族的最大似然值进化树
Figure 2 ML evolutionary tree of HD-Zip gene family between S. tenuifolia and A. thaliana and other species
图 3 荆芥HD-Zip基因家族的基因结构分析
Figure 3 Gene structure analysis of HD-Zip gene family in S. tenuifolia
图 4 荆芥HD-Zip基因家族的保守基序分析
Figure 4 Conservative motif analysis of HD-Zip gene family in S. tenuifolia
图 5 荆芥HD-Zip基因家族的顺式作用元件分布
Figure 5 Distribution of cis-acting elements of HD-Zip gene family in S. tenuifolia
图 6 荆芥HD-Zip基因家族的组内串联重复分析
Figure 6 Tandem repeat analysis of HD-Zip gene family in genome of S. tenuifolia
图 7 荆芥HD-Zip基因家族的组内共线性分析
Figure 7 Intra-group collinearity analysis of HD-Zip gene family in S. tenuifolia
图 8 荆芥HD-Zip与拟南芥基因组之间的共线性分析
Figure 8 Collinear analysis of HD-Zip gene between S. tenuifolia and A. thaliana genomes
表 1 荆芥HD-Zip基因家族的蛋白特征
Table 1. Protein characteristics of HD-Zip gene family in S. tenuifolia
亚家族 基因ID CDS长
度/bp蛋白长
度/个分子量/
kDa等电点 染色体 亚家族 基因ID CDS长
度/bp蛋白长
度/个分子量/
kDa等电点 染色体 HD-Zip Ⅰ Sch000003181 900 299 33.847 4.88 Chr 01 Sch000003831 528 175 20.331 8.58 Chr 01 Sch000016777 897 298 34.379 6.55 Chr 04 Sch000019486 822 273 31.100 4.59 Chr 04 HD-Zip Ⅲ Sch000001735 2562 853 93.306 5.74 Chr 01 Sch000026498 927 308 35.350 5.01 Chr 06 Sch000019857 2511 836 91.461 5.94 Chr 04 Sch000008725 822 273 31.034 4.83 Chr 02 Sch000014693 2493 830 91.021 5.84 Chr 03 Sch000005997 912 303 34.369 4.97 Chr 02 Sch000026324 2586 861 94.183 6.14 Chr 06 Sch000020328 966 321 35.730 4.81 Chr 04 Sch000028231 2529 842 92.434 6.17 Chr 06 Sch000016424 867 288 32.633 6.32 Chr 04 Sch000017902 879 292 33.207 6.07 Chr 04 HD-Zip Ⅳ Sch000026062 2166 721 79.071 5.98 Chr 06 Sch000002474 876 291 32.531 5.7 Chr 01 Sch000008059 2163 720 79.056 6.23 Chr 02 Sch000008983 696 231 26.832 6.32 Chr 02 Sch000029960 2181 726 79.526 5.64 HiC_scaffold_8 Sch000006831 693 230 26.615 6.96 Chr 02 Sch000011892 2418 805 87.913 5.79 Chr 03 Sch000019278 600 199 22.673 8.44 Chr 04 Sch000009216 2196 731 79.888 5.79 Chr 02 Sch000021651 546 181 21.788 5.84 Chr 05 Sch000019506 2400 799 87.073 6.04 Chr 04 Sch000013738 654 217 24.934 7.59 Chr 03 Sch000012322 2490 829 91.502 5.41 Chr 03 Sch000016710 621 206 24.472 5.44 Chr 04 Sch000012213 2490 829 91.502 5.41 Chr 03 Sch000004651 2490 829 91.502 5.41 HiC_scaffold_100 HD-Zip Ⅱ Sch000028530 891 296 327.360 7.52 Chr 06 Sch000018911 2070 689 77.061 6.28 Chr 04 Sch000010966 879 292 32.623 7.62 Chr 03 Sch000005323 1944 647 73.264 7.27 Chr 02 Sch000019272 795 264 294.950 8.59 Chr 04 Sch000022402 2307 768 84.180 5.81 Chr 05 Sch000008316 783 260 29.093 8.12 Chr 02 Sch000024046 2274 757 84.279 6.15 Chr 06 Sch000025447 879 292 32.351 9.05 Chr 06 Sch000008675 1884 627 69.905 6.18 Chr 02 Sch000011951 645 214 24.334 8.24 Chr 03 说明:CDS指蛋白编码区  下载: 导出CSV
下载: 导出CSV
-
[1] 国家药典委员会. 中华人民共和国药典 (一部)[M]. 北京: 中国医药科技出版社, 2020: 243 − 244. National Pharmacopoeia Board. Chinese Pharmacopoeia (Volume Ⅰ)[M]. Beijing: China Medical Science Press, 2020: 243 − 244. [2] LIU C, SRIVIDYA N, PARRISH A N, et al. Morphology of glandular trichomes of Japanese catnip (Schizonepeta tenuifolia Briquet) and developmental dynamics of their secretary activity [J]. Phytochemistry, 2018, 150: 23 − 30. [3] 樊佳新, 王帅, 孟宪生, 等. HPLC法测定不同产地荆芥中6种黄酮类成分[J]. 中草药, 2017, 48(11): 2292 − 2295. FAN Jiaxin, WANG Shuai, MENG Xiansheng, et al. Determination of six flavonoids in Schizonepeta tenuifolia from different areas by HPLC [J]. Chinese Traditional and Herbal Drugs, 2017, 48(11): 2292 − 2295. [4] FEDERICO DA, PABLO A M, CARLOS A D, et al. The true story of the HD-Zip family [J]. Trends in Plant Science, 2007, 12(9): 419 − 426. [5] 李媛. 大麦HD-Zip基因家族分析及功能研究[D]. 西宁: 青海大学, 2020. LI Yuan. Analysis and Functional Study of HD-Zip Gene Family in Barley [D]. Xining: Qinghai University, 2020. [6] SESSA G, CARABELLUI M, POSSENTI M, et al. Multiple links between HD-Zip proteins and hormone networks[J/OL]. International Journal of Molecular Sciences, 2018, 19(12): 4047[2022-05-04]. doi: 10.3390/ijms19124047. [7] MIYUKI N, HIROSHI K, MITSUTOMO A, et al. Characterization of the class Ⅳ homeodomain-leucine zipper gene family in Arabidopsis [J]. Plant Physiology, 2006, 141(4): 1363 − 1375. [8] BRANDT R, CABEDO M, XIE Y, et al. Homeodomain leucine-zipper proteins and their role in synchronizing growth and development with the environment [J]. Journal of Integrative Plant Biology, 2014, 56(6): 518 − 526. [9] LI Yuxia, YANG Zongran, ZHANG Yuanyuan, et al. The roles of HD-ZIP proteins in plant a biotic stress tolerance[J/OL]. Frontiers in Plant Science, 2022, 13: 1027071[2022-05-04]. doi: 10.3389/fpls.2022.1027071. [10] YUE Hong, SHU Duntao, WANG Meng, et al. Genome-wide identification and expression analysis of the HD-Zip gene family in wheat (Triticum aestivum L. )[J/OL]. Genes, 2018, 9(2): 70[2022-05-02]. doi: 10.3390/genes9020070. [11] CHEN Chengjie, CHEN Hao, ZHANG Yi, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data [J]. Molecular Plant, 2020, 13(8): 1194 − 1202. [12] MA Gang, ZELMAN A K, APICELLA P V, et al. Genome-wide identification and expression analysis of homeodomain leucine zipper subfamily Ⅳ(HD-Zip Ⅳ) gene family in Cannabis sativa L. [J/OL]. Plants. 2022, 11(10): 1307[2022-05-02]. doi: 10.3390/plants11101307. [13] ZHAO Yang, ZHOU Yuqing, JIANG Haiyan, et al. Systematic analysis of sequences and expression patterns of drought-responsive members of the HD-Zip gene family in maize[J/OL]. PLoS One, 2011, 6(12): e28488[2022-05-02]. doi: 10.1371/journal.pone.0028488. [14] WANG Zhong, WANG Shanshan, XIAO Yansong, et al. Functional characterization of a HD-Zip Ⅳ transcription factor NtHDG2 in regulating flavonols biosynthesis in Nicotiana tabacum [J]. Plant Physiology and Biochemistry, 2020, 146: 259 − 268. [15] WAN Li, DONG Jieya, CAO Minxuan, et al. Genome-wide identification and characterization of HD-Zip genes in potato [J]. Genes, 2019, 697: 103 − 117. [16] SCHRICK K, NGUYEN D, KARLOWSKI W M, et al. START lipid/sterol-binding domains are amplified in plants and are predominantly associated with homeodomain transcription factors[J/OL]. Genome Biology, 2004, 5: R41[2022-05-02]. doi: 10.1186/gb-2004-5-6-r41. [17] MUKHERIEE K, BURGLIN TR, MEKHLA, a novel domain with similarity to PAS domains, is fused to plant homeodomain-leucine zipper Ⅲ proteins[J]. Plant Physiology, 2006, 140(4): 1142 − 1150. [18] GUO Qing, JIANG Jiahui, YAO Wenjing, et al. Genome-wide analysis of poplar HD-Zip family and over-expression of PsnHDZ63 confers salt tolerance in transgenic Populus simonii × P. nigra[J/OL]. Plant Science, 2021, 311: 111021[2022-11-21]. doi: 10.1016/j.plantsci.2021.111021. [19] 邵晨冰, 黄志楠, 白雪滢, 等. 辣椒HD-Zip基因家族鉴定、系统进化及表达分析[J]. 中国农业科学, 2020, 53(5): 1004 − 1017. SHAO Chenbing, HUANG Zhinan, BAI Xueying, et al. Identification, systematic evolution and expression analysis of HD-Zip gene family in Capsicum annuum [J]. Scientia Agricultura Sinica, 2020, 53(5): 1004 − 1017. [20] NAOKO K, HITOMI O, YOSHIBUMI K, et al. Mutations in epidermis-specific HD-Zip Ⅳ genes affect floral organ identity in Arabidopsis thaliana [J]. The Plant Journal, 2013, 75(3): 430 − 440. [21] CHALVIN C, DRE VENSK S, DRON M, et al. Genetic control of glandular trichome development [J]. Trends in Plant Science, 2020, 25(5): 477 − 487. [22] YAN Tingxiang, CHEN M, SHEN Q, et al. HOMEODOMAIN PROTEIN 1 is required for jasmonate-mediated glandular trichome initiation in Artemisia annua [J]. New Phytologist, 2017, 213(3): 1145 − 1155. [23] HULSKAMP M, MISRA S, JURGENS G. Genetic dissection of trichome cell development in Arabidopsis [J]. Cell, 1994, 76(3): 555 − 566. [24] 蒋征, 王红, 吴啟南, 等. 荆芥穗药材腺鳞内含物定性及3种主要萜类的定量研究[J]. 中药材, 2016, 39(1): 31 − 36. JIANG Zheng, WANG Hong, WU Qi’nan, et al. Qualitative and quantitative analysis of major constituents of gland products in peltate glandular trichomes of Schizonepetae Spica [J]. Journal of Chinese Medicinal Materials, 2016, 39(1): 31 − 36. [25] ZHOU Peina, DANG Jingjie, SHI Zunrui, et al. Identification and characterization of a novel gene involved in glandular trichome development in Nepeta tenuifolia[J/OL]. Frontiers in Plant Science, 2022, 13: 936244[2022-05-02]. doi: 10.3389/fpls.2022.936244. -
-
链接本文:
https://zlxb.zafu.edu.cn/article/doi/10.11833/j.issn.2095-0756.20220390
 点击查看大图
点击查看大图
计量
- 文章访问数: 2967
- HTML全文浏览量: 423
- PDF下载量: 159
- 被引次数: 0
