





-
磷(P)是植物生长发育所必需的大量营养元素,对细胞组成、能量生成、新陈代谢和信号转导等关键细胞功能都具有重要作用[1]。由于土壤中磷的质量分数及流动性非常低,无机磷成为全球作物生产力的常见限制因素[2],缺磷是作物生产力损失的主要原因之一,因此,提高磷素利用效率对可持续粮食生产至关重要。为了提高磷的利用率,植物通过复杂的反应体系以改善从环境中获取磷素的途径,并在植物内循环中加以利用[3]。单纯地增加磷的吸收只能使磷从环境中输出,因此提高磷从衰老器官向幼嫩器官和发育器官的再利用效率是提高作物磷素利用效率的一个重要发展方向[4]。在许多植物中,磷脂降解是磷再活化的主要驱动因素[5]。
甘油磷酸二酯磷酸二酯酶(GDPD)将甘油磷酸二酯水解为甘油-3-磷酸和相应的醇,是甘油磷酸二酯代谢途径中的重要成员。这类基因被认为在磷酸盐稳态中发挥重要作用[6]。在原核生物和真核生物中都存在GDPD,它在各种生理过程中都发挥着重要作用[7],因此该蛋白具有生化和生物功能多样性。在哺乳动物中,GDPD与G蛋白信号转导、磷酸肌醇代谢、渗透保护、运动神经元分化、细胞骨架组织、成骨细胞分化和成肌分化有关[8]。第1个GDPD编码基因在大肠埃希菌Escherichia coli中被鉴定,它积极参与甘油和甘油-3-磷酸的摄取和代谢,因此在碳水化合物代谢和磷脂生物合成中发挥重要作用[9]。有关植物中GDPD的内容相对较少[10]。GDPD首先在磷饥饿胡萝卜Daucus carota var. sativa、美国梧桐Platanus occidentalis和拟南芥Arabidopsis thaliana细胞悬浮培养物的液泡和细胞壁中被发现[10−11]。细胞膜以磷脂的形式占细胞有机磷的三分之一,因此,在缺磷条件下从膜磷脂中再活化磷可以成为提高磷利用效率的重要策略[12]。
中国的棉花Gossypium栽种始于宋元时期,通过引进新的品种打开了一种区域化、大面积的棉花种植模式[13],此后不断引入种质资源、培育棉花品种,逐步改善了棉花的纤维品质,提高了中国棉花的生产率及效益。1978—2019年中国棉花种植面积逐年减少,中国农业部要求致力提高单产、品质,增加效益[14]。因此必须不断优化棉花品种,提升棉花品质,而缺磷是作物生产力损失的主要原因之一。本研究以陆地棉‘新陆早19’ Gossypium hirsutum ‘Xinluzao 19’为试验材料,克隆了GhGDPD1基因。采用生物信息学工具分析其编码蛋白理化性质,并对其进行表达特性分析,旨在为深入解析棉花GhGDPD1基因的生物学功能提供科学参考,并为培育磷高效利用的棉花新品种提供基因资源。
-
采用前期研究筛选出的磷高效陆地棉品种‘新陆早19’为试验材料,在河南科技大学农学院(34°59′N,112°42′E)进行大田种植,选取长势一致的‘新陆早19’植株采集根、茎、叶、花组织,用于分析GhGDPD1基因在4个组织中的表达状况。
将‘新陆早19’种子播种于含干净湿润细沙的塑料盆内,28 ℃恒温培养箱(光照14 h、黑暗10 h)培养至棉苗三叶期,选择长势良好且一致的幼苗移至水培盆中,用 1/2 Hoagland 营养液培养1周后分为2个处理水平,即适磷处理(SP: 1.00 mmol·L−1)和低磷处理(LP: 0.01 mmol·L−1),磷源为磷酸二氢钾(KH2PO4)。为保证K+的浓度一致,以1.0 mmol·L−1为标准,低磷营养液中以氯化钾(KCl)补齐K+,其他营养成分含有2.0 mmol·L−1 Ca(NO3)2·4H2O、2.5 mmol·L−1KNO3、0.5 mmol·L−1NH4NO3、1.4 mmol·L−1MgSO4·7H2O、1.0×10−3 mmol·L−1 ZnSO4·7H2O、1.0×10−3 mmol·L−1MnSO4·H2O、1.0×10−4 mmol·L−1CuSO4·5H2O、 1.0×10−2 mmol·L−1H3BO3、0.5×10−5 mmol·L−1 (NH4)6MO7O24·4H2O、0.1 mmol·L−1EDTA-FeNa[15]。分别处理0、4、12、24、72 h后,选取3株生长一致的‘新陆早19’植株,并混合取其根部组织,然后迅速置于液氮中,−80 ℃保存备用。每日调节营养液 ,使pH保持在 6.5 左右,每3 d更换1次营养液。
-
根据前期陆地棉根部低磷胁迫基因表达谱芯片差异表达基因序列进行分析,在美国国家生物技术信息中心(NCBI)网站的EST数据库中检索差异表达基因序列,得到该基因的相似序列。将所得相似性序列(覆盖率>50%,相似度>90%)使用DNASTAR的Seqman进行拼接得到重叠群(conting),将所得序列继续检索与拼接直至没有新的相似序列出现,所得即为结果序列重叠群,利用ORFfinder在线平台查找开放阅读框,进行目标基因GhGDPD1的克隆与分析。
根据ORFfinder在线平台查找的开放阅读框,使用Primer 5.0软件设计引物,由生工生物工程有限公司合成(表1)。
引物 引物序列(5′→3′) 引物 引物序列(5′→3′) Sense primer ATTTTTCCCTCTCTTACTCTATCCC GhActin-F ATCCTCCGTCTTGACCTTG Anti-sense primer GTAGGGACAAGTTAGTGGTGTATCA GhActin-R TGTCCGTCAGGCAACTCAT 通用引物M13F TGTAAAACGACGGCCAGT GhGDPD1-F TTCTCTGTCTCTCTACTCGTCTCGT 通用引物M13R CAGGAAACAGCTATGACC GhGDPD1-R TCTATGCCCTATTACCAAAAACTTC Table 1. Primers used in the study
-
取液氮速冻整株‘新陆早19’植株在研钵中迅速研磨成粉末转移至离心管,利用CTAB法提取‘新陆早19’的基因组DNA,向其中添加200 μL的TE缓冲液溶解后置于−20 ℃下保存备用。
以‘新陆早19’的基因组DNA为模板设计如下PCR扩增反应体系(20.0 μL)。冰上操作:2×M5 HiPer plus Taq HiFi PCR mix (with blue dye) 10.0 μL,Sense primer (10.0 μmol·L−1) 0.5 μL,Anti-sense primer (10.0 μmol·L−1) 0.5 μL,Template DNA 0.5 μL,Nuclease-free ddH2O 8.5 μL。反应程序为:95 ℃ 3 min;94 ℃ 25 s,55 ℃ 25 s,72 ℃ 50 s,35个循环;72 ℃ 5 min;−4 ℃低温保存。120 V,25 min,质量分数为1%琼脂糖凝胶电泳检测。
将目的DNA片段进行胶回收纯化后连接pTOPO-T载体,具体体系为(5.0 μL):M5 HiPer pTOPO-TA Vector 0.5 μL,10×Enhancer 0.5 μL,纯化后的PCR产物 1.8 μL,灭菌水 2.2 μL。取5.0 μL连接产物转化50.0 μL大肠埃希菌Escherichia coli DH5ɑ感受态细胞,将菌液涂布在含氨苄青霉素的LB固体培养基上,37 ℃培养过夜。随机挑取单菌落,扩大培养4 h。以所得菌液作模板进行菌液PCR反应,体系如下(20.0 μL):2× M5 HiPer plus Taq HiFi PCR mix (with blue dye) 10.0 μL,通用引物M13F 0.5 μL,通用引物M13R 0.5 μL,Template DNA 0.5 μL,Nuclease-free ddH2O 8.5 μL。反应程序为:95 ℃ 3 min;94 ℃ 25 s,55 ℃ 25 s,72 ℃ 50 s,35个循环;72 ℃ 5 min;−4 ℃低温保存。检测符合后送至生工生物工程有限公司测序。
-
取液氮速冻整株‘新陆早19’植株在研钵中迅速充分研磨成粉末,后续步骤依照所用试剂盒说明书进行,150 V,15 min,质量分数为1%琼脂糖凝胶电泳检测。参考试剂盒说明书完成cDNA的合成,−20 ℃保存备用。
以‘新陆早19’的cDNA为模板设计如下PCR扩增反应体系(20.0 μL)。冰上操作:2×M5 HiPer plus Taq HiFi PCR mix (with blue dye) 10.0 μL,Sense primer (10.0 μmol·L−1) 0.5 μL,Anti-sense primer (10.0 μmol·L−1) 0.5 μL,cDNA 1.0 μL,Nuclease-free ddH2O 8.0 μL。PCR反应程序为:95 ℃ 3 min;94 ℃ 25 s,56 ℃ 25 s,72 ℃ 45 s,35个循环;72 ℃ 5 min;−4 ℃低温保存。120 V,25 min,质量分数为1%琼脂糖凝胶电泳检测。
将目的DNA片段进行胶回收纯化后连接pTOPO-T载体,体系如下(5.0 μL):M5 HiPer pTOPO-TA Vector 0.5 μL,10×Enhancer 0.5 μL,纯化后的PCR产物 1.5 μL,灭菌水 2.5 μL。加入50.0 μL大肠埃希菌DH5ɑ感受态细胞转化,挑菌进行菌液PCR反应。电泳检测符合后送至生工生物工程有限公司进行测序。
-
使用在线平台及软件对基因进行生物信息学分析,预测分析基因的结构、性质等。
-
利用半定量RT-PCR技术,分析GhGDPD1基因在根、茎、叶、花4个组织中的表达状况。以‘新陆早19’各组织的cDNA为模板,GhActin作内参基因,所用引物见表1。设计如下PCR扩增反应体系(20.0 μL)。冰上操作:2×M5 HiPer plus Taq HiFi PCR mix (with blue dye) 10.0 μL,Sense primer (10.0 μmol·L−1) 0.5 μL,Anti-sense primer (10.0 μmol·L−1) 0.5 μL,cDNA 1.0 μL,Nuclease-free ddH2O 8.0 μL。PCR反应程序为:95 ℃ 3 min;94 ℃ 25 s,60 ℃ 25 s,72 ℃ 10 s,35个循环;72 ℃ 5 min;−4 ℃低温保存。120 V,25 min,质量分数为1%琼脂糖凝胶电泳检测。
-
采用RT-qPCR技术检测GhGDPD1基因在不同组织中及低磷胁迫处理下的表达模式。以GhActin作为内参基因,所用引物见表1。采用SYBR® Green Pro Taq HS预混型qPCR试剂盒进行荧光定量,使用的荧光定量PCR仪器型号是CFX96,数据分析采用2−ΔΔCt法。
-
以实验室前期低磷胁迫差异表达序列为探针,检索发现并下载了10条相似序列,利用DNASTAR将所得序列全部拼接得到新的重叠群,对新得到的序列再次进行检索,在第1次检索序列的基础上又增加了1条新的序列,将11条基因序列全部拼接,得到了1个新的重叠群,序列长度为1 462 bp。
-
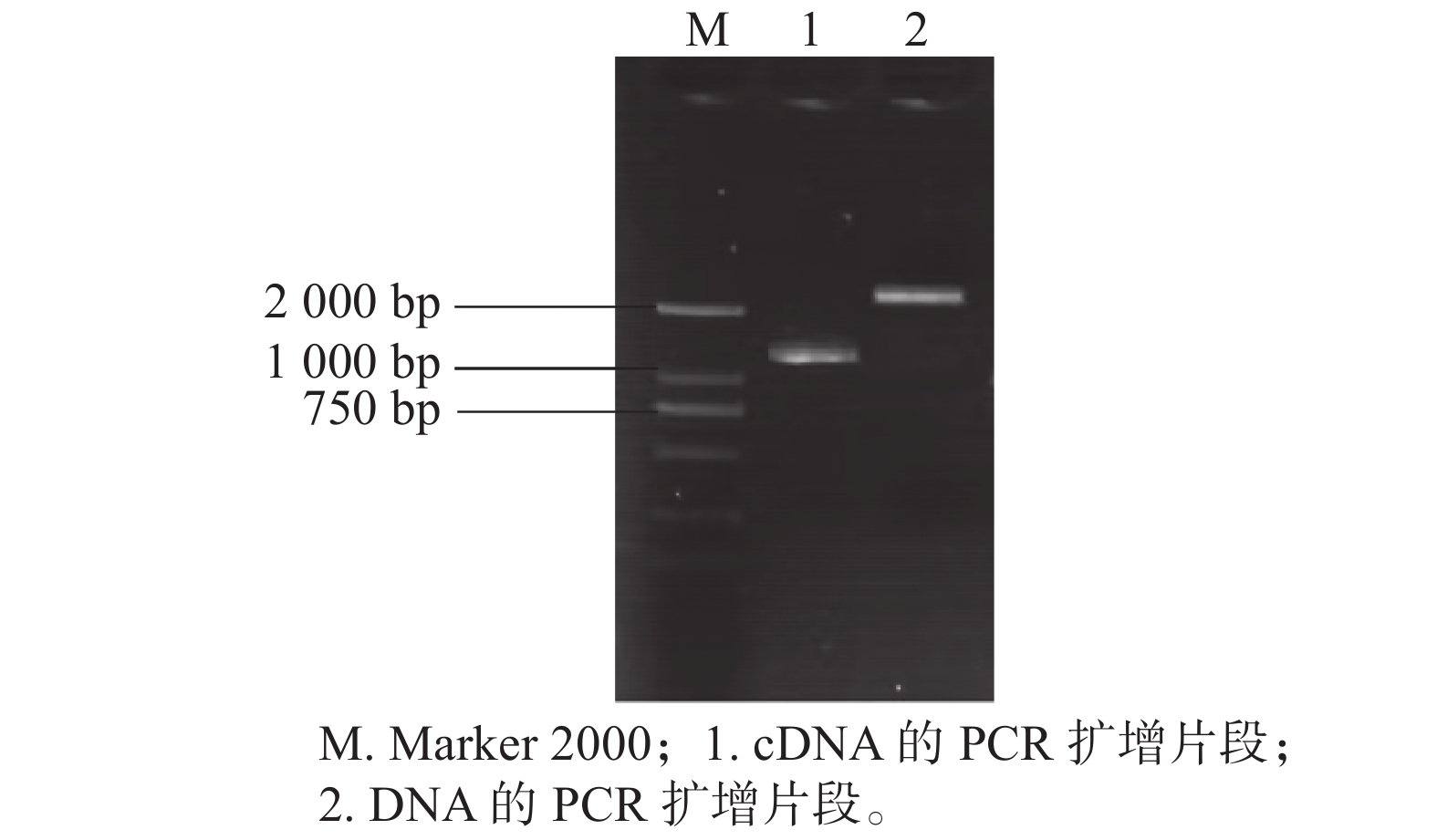
根据得到的克隆重叠群,横跨GhGDPD1基因的编码序列(coding sequence,CDS)区设计特异性引物,以‘新陆早19’植株的DNA和cDNA为模板,克隆得到GhGDPD1基因的CDS (图1),其条带符合目的条带大小。通过无缝克隆连接到pTOPO-T载体上,挑选阳性克隆测序。获得的DNA序列大于2 000 bp (图1,泳道2),与重叠群序列的一致性较高,均大于89%。获得cDNA序列1 200 bp (图1,泳道1),与重叠群序列比对一致性为99%。开放阅读框为长度为1 149 bp,共编码382个氨基酸。该编码区具有GDPD_GDE5_like_1_plant结构域,因此GhGDPD1基因是GDPD家族成员。

Figure 1. Cloning of GhGDPD1 CDS
-
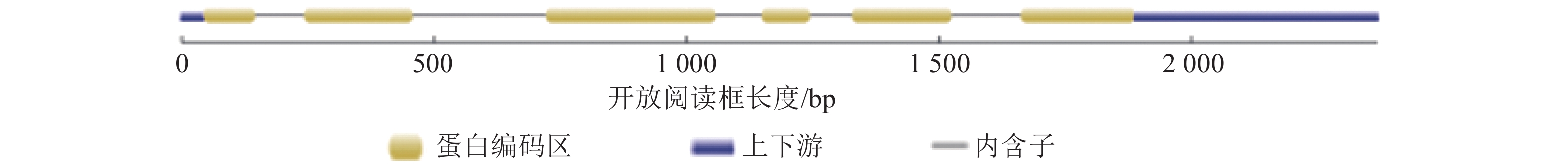
将DNA测序结果序列与cDNA测序结果的最大开放阅读框序列导入Gene Structure Display Server 2.0,对该基因组序列与编码序列进行分析,表明该基因编码序列包含5′与3′非编码序列,共有5个内含子,6个外显子(图2)。

Figure 2. GhGDPD1 gene structure analysis
-
将基因编码蛋白导入ExPASy-ProtParam tool在线软件,分析可知:该基因共编码了382个氨基酸,所编码蛋白的分子式为C1930H3030N506O580S12,脂肪族氨基酸指数为94.84,分子量为42 987.05 Da,等电点为5.17,属于酸性蛋白。其中亮氨酸的相对含量占比最大,为10.2%,共39个。在该蛋白中带正电荷的氨基酸有38个,带负电荷的有54个。氨基酸组分会直接影响蛋白的亲疏水性,总亲水性平均系数为−0.179,不稳定系数为41.79,是亲水性不稳定蛋白。ProtScale analysis在线软件显示:此蛋白中精氨酸亲水性最强,亲水指数为−4.500 0,异亮氨酸疏水性最强,亲水指数为4.500 0。
-
在NPS@:SOPMA secondary structure prediction网站预测该蛋白二级结构,其中占比最大的α-螺旋占40.84%,包含156个氨基酸,其次是无规则卷曲占比为38.22%,包含146个氨基酸。此外,此蛋白还含有延伸链(extended strand)和β-转角(beta turn),其中延伸链占比较多,为15.18%,有58个氨基酸,β-转角仅有22个氨基酸,占比为5.76%,是最少的。表明此蛋白的二级结构由α-螺旋和无规则卷曲占据主体地位所组成,各结构在各个区间分布较为均匀。由于无规则卷曲占比较多,预测其结构比较复杂。
运用TMHMM-2.0分析发现:该基因所编码蛋白不含有跨膜结构域,属于非跨膜蛋白,382个氨基酸均位于细胞膜表面,并未形成跨膜螺旋区。通过SignalP-5.0进行信号肽分析,可知:GhGDPD1基因编码蛋白有信号肽的概率是0.000 3,其他的可能性则高达0.999 7。382个氨基酸中并未出现典型的信号肽趋势,即该蛋白不存在信号肽。
在Plant-mPLoc在线网站内输入基因编码序列进行基因编码蛋白的亚细胞定位预测,预测定位在细胞膜(Cell membrane)。预测结果与跨膜结构预测分析结果相同,均为该蛋白位于膜外,推测该基因可能直接在细胞膜发挥作用。
-
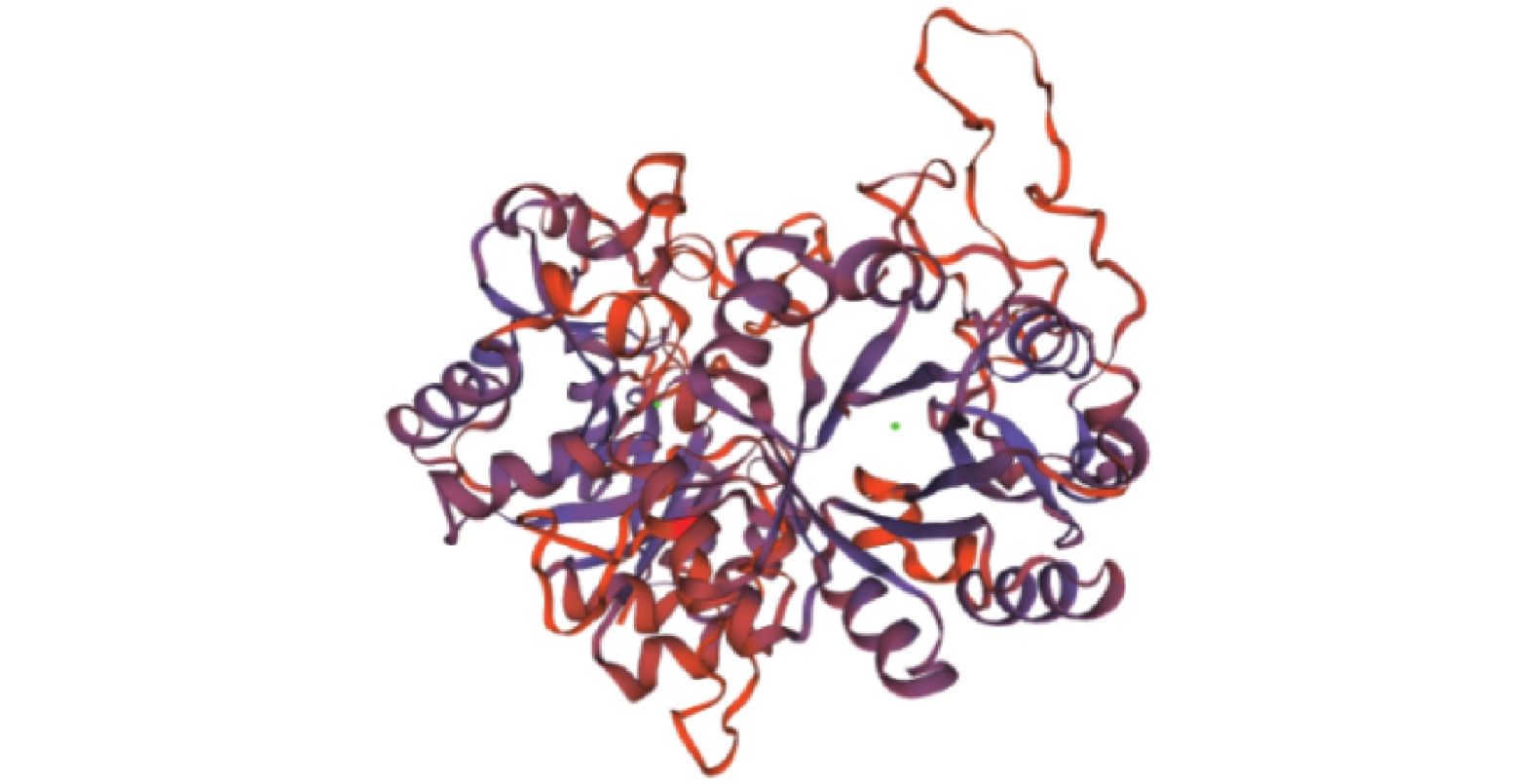
通过SWISS-MODEL Interactive Workspace进行同源建模,预测蛋白三级结构(图3)。评价同源建模 (QMEAN)结果为0.57,全球性模型质量估测 (GMQE)值为0.44,预测其含有甘油磷酸二酯磷酸二酯酶,与腾冲嗜热菌Thermoanaerobacter tengcongensis MB4的甘油磷酸二酯磷酸二酯酶的晶体结构类似。蛋白的三级结构与二级结构分析结果一致,均以α螺旋和无规则卷曲为主体。
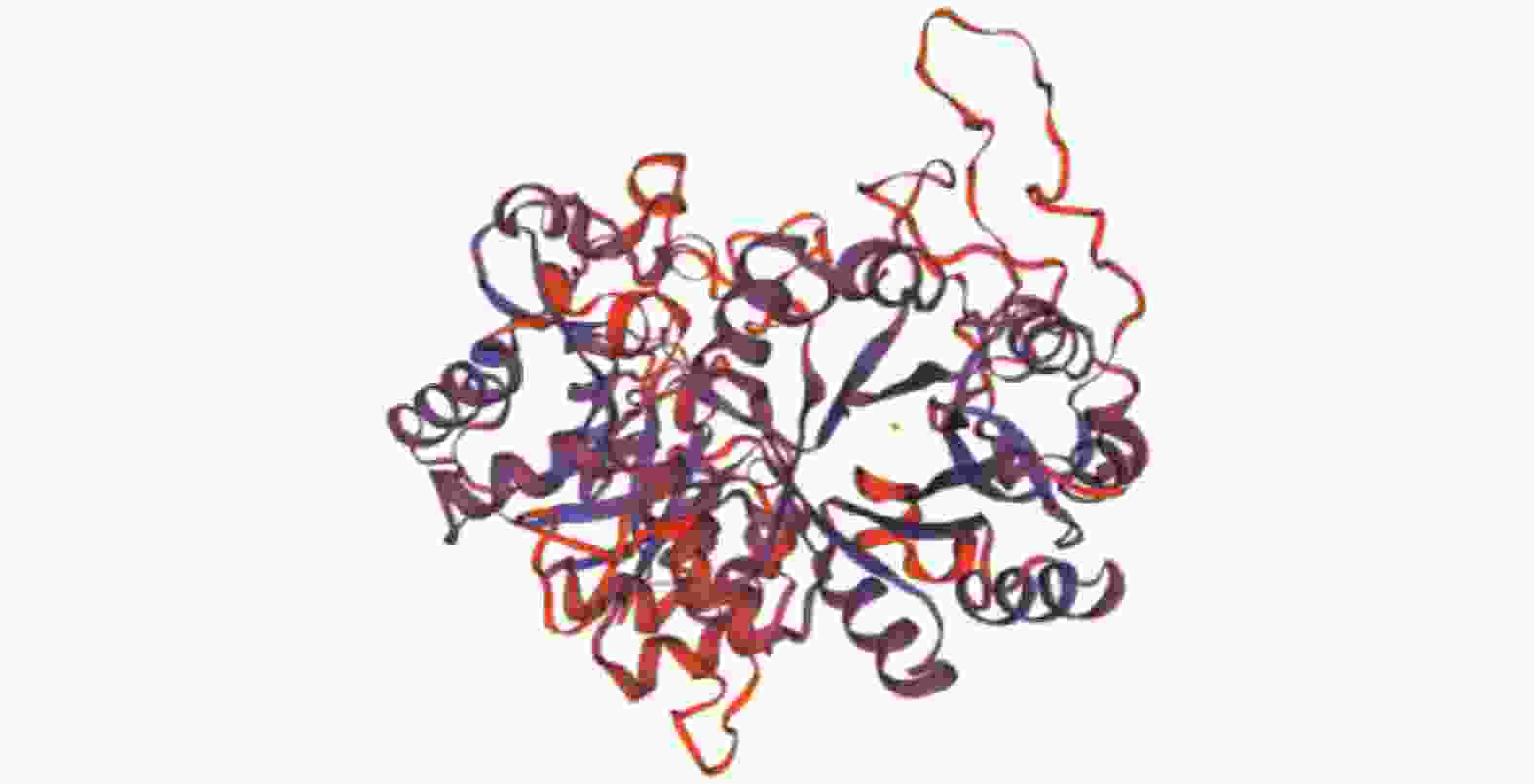
Figure 3. Predicted three-dimension structure of GhGDPD1 protein
将获得的基因编码序列提交到STRING 11.0在线网站,预测该基因可能的互作网络。分析结果显示:GhGDPD1基因编码蛋白能够预测到蛋白质的相互作用网络,因此预测GhGDPD1基因可能是通过多个蛋白互作进行调控。
N-糖基化对蛋白稳定及功用均会产生极其重要的影响。将获得的基因编码序列提交到NetNGlyc 1.0在线软件,可知该蛋白存在3个潜在的N-糖基化位点(分别位于7~10、21~24、116~119氨基酸区段)。磷酸化位点与蛋白的功能、结构都有很大的关系。通过NetPhos 3.1在线网站分析可知:此蛋白中占比最大的是丝氨酸和苏氨酸磷酸化位点。GhGDPD蛋白除了可以调控蛋白的活性以外,在细胞的信号转导中也占据了重要的位置。
-
使用NCBI的保守结构域数据库(CDD-search)查找GhGDPD1氨基酸序列的保守结构域。分析结果显示:GhGDPD1基因所编码蛋白含有1个特异匹配,为GDPD_GDE5_like_1_plant,匹配所属的超家族是PI-PLCc_GDPD_SF基因家族。GhGDPD1基因与其他物种的GDPD1氨基酸序列进行多重比对分析,同源性较高,物种间GDPD蛋白氨基酸长度基本一致,同源性达84.12%(图4)。

Figure 4. Multi-alignment of GhGDPD1 amino acid sequence with other GDPD family in different plants
在NCBI数据库利用BLASTp检索氨基酸序列的同源序列,下载了榴莲Durio zibethinues、木槿Hibiscus syriacus、可可树Theobroma cacao、哥伦比亚锦葵Herrania umbratica、长蒴黄麻Corchorus olitorius、麻疯树Jatropha curcas、芒果Mangifera indica、胡杨Populus euphratica、开心果Pistacia vera、白梨Pyrus bretschneideri、金钱橘Citrus clementina等11条不同物种的蛋白序列。通过软件MEGA 5构建系统进化树(图5),‘新陆早19’蛋白序列与木槿的亲缘关系最近,其次与可可树、哥伦比亚锦葵、榴莲、长蒴黄麻的亲缘关系较近,与其余6种物种的亲缘关系较远,不在一个大支上。

Figure 5. GhGDPD1 phylogenetic tree analysis
-
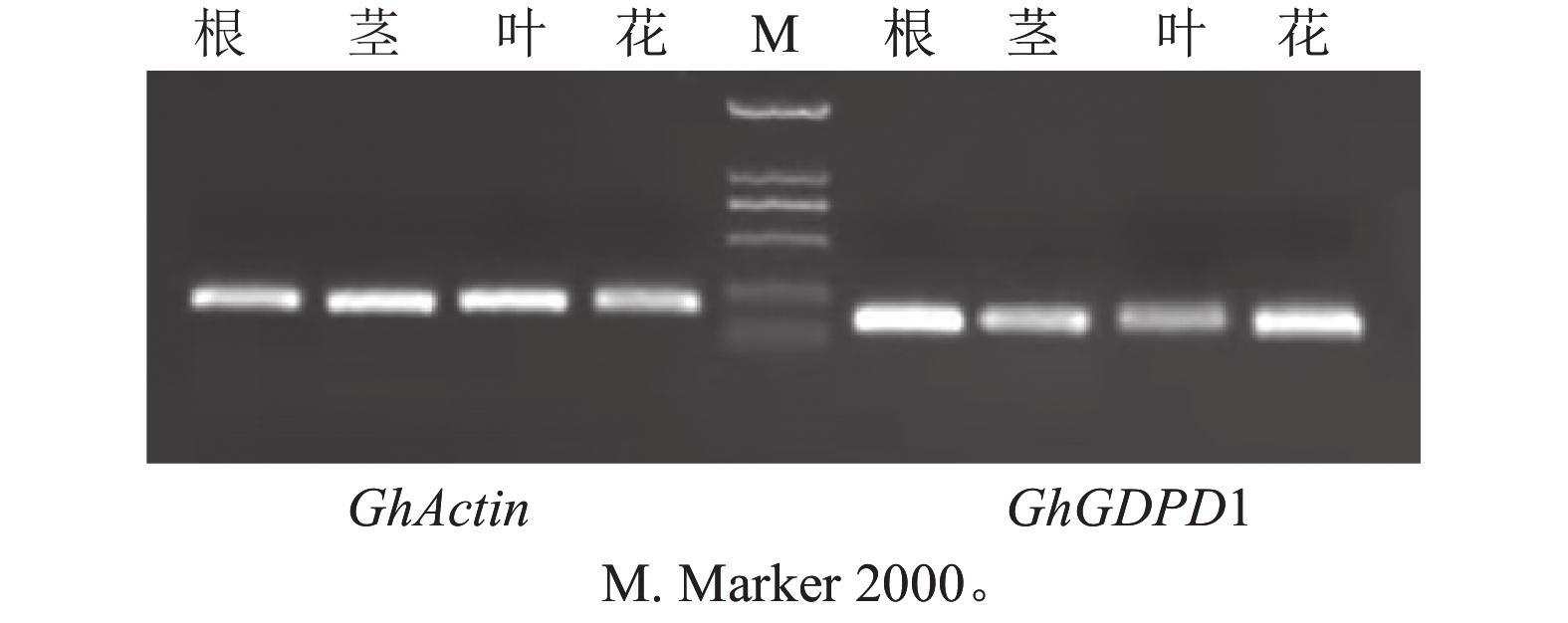
以棉花的GhActin为内参基因,通过半定量RT-PCR技术对GhGDPD1基因在‘新陆早19’的4个组织中的表达情况进行分析。从图6可知:GhGDPD1高表达于根,微量表达于叶,中量表达于茎和花。利用RT-qPCR检测GhGDPD1基因在‘新陆早19’植株的根、茎、叶、花4个组织中的表达结果(图7A),结果与半定量RT-PCR分析一致。由图7B可以看出:在低磷处理4~24 h时相对表达量逐渐下降,但相对表达量均比适磷情况下高,在72 h时其相对表达量又上升,仅略低于4 h时的相对表达量。其中,4、12、72 h时低磷处理比适磷处理显著表达(P<0.05),说明GhGDPD1基因在受到低磷胁迫刺激后,会立即对低磷胁迫做出应答,且具有持续性。

Figure 6. Expression of GhGDPD1 in different tissues of cotton by semi-quantitative RT-PCR results
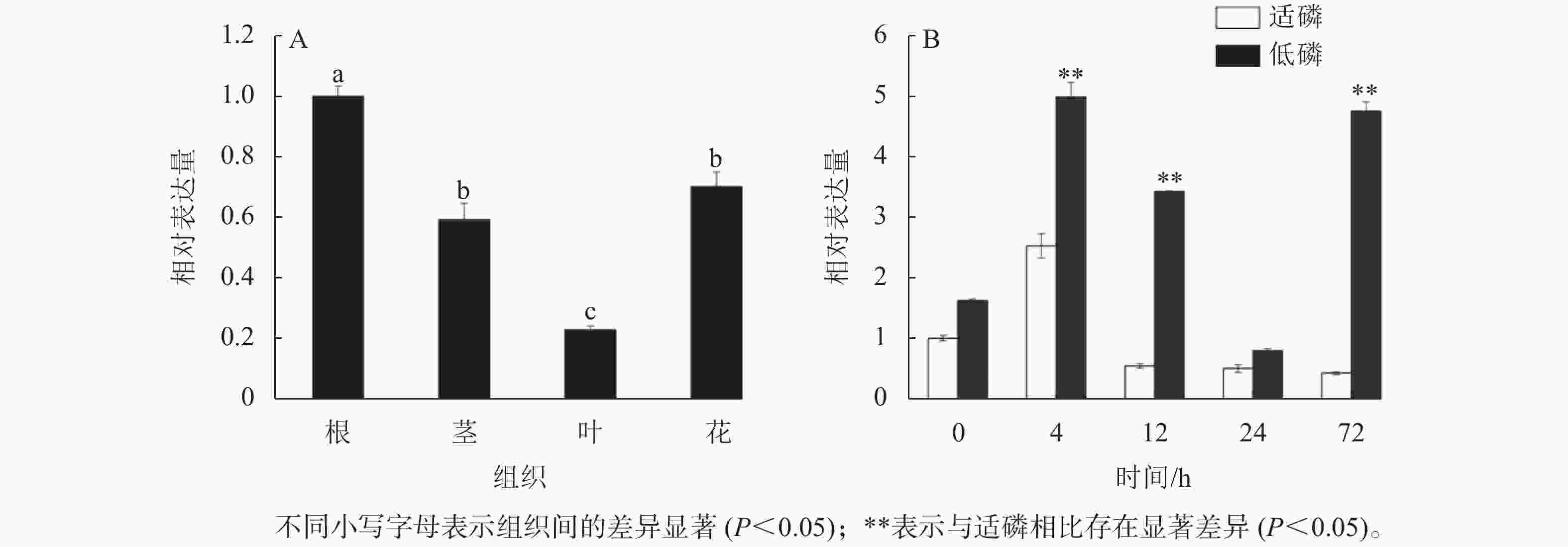
Figure 7. Expression analysis of GhMGD1 in different tissues (A) and low phosphorus stress (B)
-
磷是植物最重要的必需营养素之一,因为它是核酸、核苷酸和磷脂等关键大分子的组成部分,并且以磷酸盐或磷酯的形式参与新陈代谢和蛋白质调节。但磷的特性也导致植物对磷肥的低效利用。施加的磷肥只有15%~25%被植物吸收,其余的则渗入环境,造成土壤退化和水体富营养化等有害后果[16]。GDPD已被鉴定为拟南芥中的多基因家族[17]。所有已鉴定的拟南芥GDPD被分为2组:具有1个GDPD结构域的A型GDPD和具有2个推定的GDPD结构域的B型GDPD。其中B型GDPD具有植物特异性,磷酸二酯酶活性相对较低。据报道,GDPD基因能水解甘油磷酸二酯,并在磷饥饿时显著诱导,表明它们在低磷胁迫中存在潜在作用[18]。
本研究以陆地棉‘新陆早19’植株的cDNA为模板克隆到1个GDPD1基因。该基因序列开放阅读框的长度为1 149 bp,共编码382个氨基酸;该基因的结构比较复杂,有5个内含子,1个GDPD_GDE5_like_1_plant结构域,命名为GhGDPD1。生物信息学分析结果显示:GhGDPD1蛋白相对分子质量是42 987.05 Da,理论等电点为5.17,是酸性蛋白,具备不稳定性与亲水性。GhGDPD1蛋白的二级结构由α-螺旋和无规则卷曲占据主体地位所组成。通过同源建模可知:GhGDPD1基因三级结构预测其含有甘油磷酸二酯磷酸二酯酶。GhGDPD1基因编码蛋白不存在信号肽和跨膜结构域。GhGDPD1蛋白存在3个潜在的N-糖基化位点和多个酸化位点。亚细胞定位结果显示:GhGDPD1基因编码蛋白在细胞膜中,预测其能够通过蛋白质的相互作用参与调控。RT-qPCR与半定量RT-PCR结果表明:GhGDPD1基因在‘新陆早19’根中的表达量最大,在叶片中表达量最小,中量表达于花和茎。在受到低磷胁迫的刺激后,GhGDPD1基因会立即对低磷胁迫做出应答。GhGDPD1基因可能在根系发挥作用,在‘新陆早19’的磷高效利用信号调控过程中具有重要的作用。
Cloning and expression analysis of low phosphorus stress response gene GhGDPD1 in Gossypium hirsutum
doi: 10.11833/j.issn.2095-0756.20220624
- Received Date: 2022-09-28
- Accepted Date: 2023-03-24
- Rev Recd Date: 2023-03-22
- Available Online: 2023-07-13
- Publish Date: 2023-08-20
-
Key words:
- Gossypium hirsutum /
- low phosphorus stress /
- gene cloning /
- bioinformatics analysis /
- expression analysis
Abstract:
| Citation: | MENG Chaomin, GENG Feifei, QING Guixia, et al. Cloning and expression analysis of low phosphorus stress response gene GhGDPD1 in Gossypium hirsutum[J]. Journal of Zhejiang A&F University, 2023, 40(4): 723-730. DOI: 10.11833/j.issn.2095-0756.20220624

|



 DownLoad:
DownLoad: